
Abstract
Gliomas are the most commonly occurring malignant brain tumor characterized by an immunosuppressive microenvironment accompanied by profound epigenetic changes, thus influencing the prognosis. Glutathione peroxidase 7 (GPX7) is essential for regulating reactive oxygen species homeostasis under oxidative stress. However, little is known about the function of GPX7 in gliomas. In this study, we hypothesized that GPX7 methylation status could influence biological functions and local immune responses that ultimately impact prognosis in adult gliomas. We conducted an integrated bioinformatics analysis mining GPX7 DNA methylation status, transcriptional and survival data of glioma patients. We discovered that GPX7 was remarkably increased in glioma tissues and cell lines, and was associated with poor prognosis. This upregulation was significantly linked to clinicopathological and molecular features, besides being expressed in a cell cycle-dependent manner. Our results consistently demonstrated that upregulation of GPX7 is tightly modulated by epigenetic processes, which also impacted the overall survival of patients with low-grade gliomas (LGG). Based on the analysis of biological functions, we found that GPX7 might be involved in immune mechanisms involving both innate and adaptive immunity, type I interferon production and regulation of synaptic transmission in LGG, whereas in GBM, it is mainly related to metabolic regulation of mitochondrial dynamics. We also found that GPX7 strongly correlates with immune cell infiltration and diverse immune cell markers, suggesting its role in tumor-specific immune response and in regulating the migration of immune cell types to the tumor microenvironment. Combining these multiple data, we provided the first evidence regarding the epigenetic-mediated regulatory mechanisms underlying GPX7 activation in gliomas. Furthermore, our study brings key insights into the significant effect of GPX7 in modulating both immune molecules and in immune cell infiltration in the microenvironment of gliomas, which might impact the patient outcome, opening up future opportunities to regulate the local immune response.
Similar content being viewed by others


Introduction
Gliomas are the most common primary intracranial tumor, representing up to 80% of all brain tumors1. They have aggressive behavior with a high mortality rate (median survival about 15 months), and exhibit notable variability at the histopathological and molecular level. According to the World Health Organization (WHO), they are categorized by the grade of malignancy (Grades 1, 2, 3 and 4), molecular markers (e.g., IDH1/2 mutations, TERT mutations, 1p/19q co-deletion) and pathological features. As a result of an integrative view, gliomas are further segregated into 6 different families: (i) Adult-type diffuse gliomas (e.g., glioblastoma, IDH-wildtype); (ii) Pediatric-type diffuse low-grade gliomas (which have good prognosis); (iii) Pediatric-type diffuse high-grade gliomas (that behave aggressively); (iv) Circumscribed astrocytic gliomas; (v) Glioneuronal and neuronal tumors; and (vi) Ependymomas. Additionally, new molecular biomarkers have gained importance in providing both ancillary and defining diagnostic information, impacting clinical decision making and refining the WHO classification of an increasing number of gliomas2,3. As an example, the last update of WHO classification (5th edition) incorporated (i) CDKN2A/B homozygous deletion in IDH-mutant astrocytomas (covering grades 2–4), (ii) TERT promoter mutation, EGFR amplification, and + 7/− 10 copy number changes in IDH-wildtype diffuse astrocytomas as well as (iii) MYB- or MYBL1 mutations in diffuse astrocytoma. However, despite the extensive molecular characterization, they remain incurable and reliable biomarkers are needed to further elucidate the molecular mechanism of glioma development4.
The endoplasmic reticulum-localized glutathione peroxidase 7 (GPX7), also known as non-selenocysteine phospholipid hydroperoxide glutathione peroxidase (NPHGPx), is a key PDI (protein disulfide isomerase) oxidase that uses H2O2 as a source of oxidative power5. GPX7 acts as a critical intracellular sensor that detects redox level and transmits reactive oxygen species (ROS) signals to its interacting proteins (redox-sensitive thiol-containing proteins) by disulfide bonds shuttling, supporting multiple biologic processes such as oxidative protein folding5,6,7, the release of the non-targeting short interfering RNAs (siRNAs)-associated stress8,9 and protection of the organism against systemic oxidative stress5,10,11,12,13. Among the GPX7-interacting proteins, well-known examples include GRP78, CPEB2, XRN2, ADF, GRP75, HSP7C, ERp72, eEF1A-1, U-Tmod (TMOD3), ErJ3, and Histone H1b8,14. Some studies have generated compelling evidence for the relevance of GPX7 in metabolic diseases12,15,16, neurodegeneration17,18,19, viral infection20 and cardiovascular diseases8. In addition, dysregulation of GPX7 has been found to contribute in different ways to the tumorigenesis and progression of many human carcinomas, such as esophageal adenocarcinoma21, gastric cancer22, hepatocellular carcinoma23, acute myeloid leukemia24 and breast cancer25. On the other hand, the role of GPX7 in the development of gliomas and the epigenetic mechanisms underlying this process have yet to be fully characterized. To address this issue, in the present study, we used database research and bioinformatic analyses to assess the expression of GPX7 in gliomas and analyze its epigenetic modulation, potential biological functions, prognostic value and correlation with tumor-infiltrating immune cells.
Material and methods
Oncomine database analysis
Using the Oncomine database (https://www.oncomine.org)26, we examined the expression differences of GPX7 expression across several human tumors and the corresponding normal tissues. The threshold for all analyses was defined as follows: Fold change = 2.5; Gene ranking: top 10%; Data type: mRNA, and “Analysis type”: cancer vs. normal analysis. All statistical methods and values were obtained directly from the corresponding database. The student’s t-test was used to generate the p-value for expression differences and p < 0.05 was considered statistically significant. All microarray platforms were considered.
Differential gene expression analysis
To validate our findings of the differential expression found for glioma tumors in the Oncomine database, we analyzed GPX7 expression using The Cancer Genome Atlas (TCGA) RNA-seq data (LGG = 518 samples; GBM = 163 samples; Genotype-Tissue Expression (GTEx) dataset as a control dataset, N = 207 samples), Chinese Glioma Genome Atlas (CGGA) RNA-seq data (Normal samples = 20 samples; LGG = 426 samples; GBM = 225 samples), Rembrandt microarray dataset27 (Normal samples = 28 samples; LGG samples = 225 samples; GBM samples = 219 samples), Gravendeel microarray dataset28 (Normal samples = 8 samples; LGG = 117 samples; GBM = 159 samples), and Kamoun microarray cohort29 (Normal samples = 9 samples; LGG = 154 samples; GBM = 16 samples). Clinical and molecular information was downloaded from CGGA (http://www.cgga.org.cn)30, GlioVis (http://gliovis.bioinfo.cnio.es/)31 and GDC (Genomic Data Commons).
Cancer cell line encyclopedia database analysis
We expanded our analysis by evaluating the GPX7 expression in a panel with multiple human cancer cell lines (including 66 glioma cell lines) using data from the Cancer Cell Line Encyclopedia (CCLE, https://portals.broadinstitute.org/ccle/)32,33, which spans multidimensional arrays and RNA-seq datasets for over 1457 human cancer cell lines. The expression was ranked using Affymetrix GeneChip data.
Analysis of gene expression omnibus (GEO) microarray datasets
The Gene Expression Omnibus (GEO) (https://www.ncbi.nlm.nih.gov/gds)34 is a public database of the National Center of Biotechnology Information that stores high-throughput gene expression datasets. To profile the variation of GPX7 expression during the cell cycle in T98G cells (Human GBM Cell line), we downloaded and analyzed the GSE8537 dataset35, which was based on the Affymetrix GPL570 platform ([HG-U133-Plus2] Human Genome U133 Plus 2.0 Array).
Also, we obtained GPX7 gene expression profiling from three therapeutically relevant transcriptome microarrays: (i) GSE43452 dataset36, from a human GBM cell line (U87MG) treated with temozolomide at 20 µM for 24 h, based on the Illumina GPL10558 (HumanHT-12 V4.0 expression beadchip); (ii) GSE39223 dataset37, that included U87MG-EV human glioblastoma xenograft tumor treated with bevacizumab (10 mg/kg of body weight), which was subcutaneously implanted into 7 to 8-week-old female BALB/c SCID mice (Harlan Sprague Dawley, Inc., Indiana), 100μL of cell suspension mixed with an equal volume of matrigel (BD Bioscience). After 3 days, the second dose of Bevacizumab was given and, 4 h later, mice were sacrificed and the tumors were collected. This dataset was based on the Affymetrix GPL70 platform ([HG-U133-Plus2] Human Genome U133 Plus 2.0 Array); and (iii) GDS2428 dataset38, from short-term cultured glioblastoma cells (GLI56) treated with 5-aza-2′-deoxycytidine (5-aza-dC) 5 μM for 96 h to induce DNA demethylation.
UCSC (University of California Santa Cruz) Xena Browser
Heat map of the integrated analysis with clinical information (Histology and WHO grade), copy number alterations (CNAs) and expression levels of GPX7 were obtained by mining TCGA-LGG-GBM dataset (1153 samples) by using the UCSC Xena Browser (https://xena.ucsc.edu/). The gene expression profile from RNAseq data was indicated as normalized_log2[norm_count + 1] and CNAs status as Log2 (Tumor/Normal).
Clinical correlations and survival analysis
To perform the correlation between GPX7 expression and clinicopathological/molecular parameters, gene expression of normalized and pre-processed data (both microarray and RNA-seq) of the TCGA-LGG-GBM dataset and CGGA-LGG-GBM was downloaded from GlioVis (http://gliovis.bioinfo.cnio.es/)31. The latest corresponding clinical data were downloaded from GDC (Genomic Data Commons), and patients with unavailable clinical information were excluded. All statistical analyses were performed by R v4.0.5 (https://www.r-project.org/).
The disease-free survival (DFS) and overall survival (OS) curves for TCGA-LGG and TCGA-GBM cohorts and clinicopathological subgroups were conducted using the GEPIA tool (http://gepia.cancer-pku.cn)39,40. Patients were dichotomized into two groups (Red: high expression; Blue: low expression) according to the median expression level of GPX7. All survival curves were generated by the Kaplan–Meier methods. The log-rank P-value and hazard ratio (HR) with 95% confidence intervals were also calculated. The survival findings were further validated in two different glioma cohorts: (i) Chinese Glioma Genome Atlas (CGGA) (http://www.cgga.org.cn/) (n = 325 samples)41; and (ii) Repository of Molecular Brain Neoplasia Data (Rembrandt) (n = 444 samples)42,43,44.
Finally, ROC Plotter (http://www.rocplot.org/)45 was used to assess the sensitivity and specificity of GPX7 for classifying chemotherapy responsiveness in GBM patients (n = 454). We checked all included GBM patients receiving “any chemotherapy” as the primary analysis with the secondary analysis for: Bevacizumab, Irinotecan, Nitrosoureas (Lomustine, Carmustine, Estramustine, Laromustine, Nimustine), Topoisomerase inhibitor, Angiogenesis inhibitor, Carmustine, Lomustine and Temozolomide. A box plot displaying GPX7 expression levels in responders and non-responders (based on OS at 16 months) was provided along with the area under the curve (AUC) and their respective p-values. The differences were assessed with the Mann–Whitney U test. No additional filter was applied, and no outliers were excluded from the analysis.
GO enrichment analysis of GPX7-related genes in LGG and GBM based on the LinkedOmics database
To evaluate the potential functional mechanism of GPX7 in gliomas, we obtained all genes co-expressed with GPX7 from the LinkedOmics (http://www.linkedomics.org/)46,47, using the LinkFinder module. Spearman’s test was conducted to perform statistical analyses. To derive biological insights from the association results, the gene ontology (GO) enrichment analysis was conducted by Gene Set Enrichment Analysis (GSEA) with a minimum number of genes (size) of 10 and the simulation of 1000. A p-value < 0.05 was deemed to indicate statistical significance.
GPX7 Methylation analysis
The methylation levels (β-value) of CpG sites associated with GPX7 in TCGA-LGG and TCGA-GBM were assessed using the bioinformatics platform MethSurv (https://biit.cs.ut.ee/methsurv/)48. Moreover, the overall survival (OS) analysis for each CpG site was assessed using Kaplan–Meier plots. Log‐rank tests were used to measure the statistical significance and Log‐rank p < 0.05 was considered significant. The methylation pattern of each probe indicating subregions of the query gene was plotted using a heatmap by ClustVis (https://biit.cs.ut.ee/clustvis/)49.
Thereafter, Shiny Methylation Analysis Resource Tool (SMART; http://www.bioinfo-zs.com/smartapp/)50 was used to determine the correlation between the degree methylation of each probe (β-value) and the corresponding expression level of GPX7 in both TCGA-LGG and TCGA-GBM cohorts. The threshold of all analyses was determined as follows: Aggregation method: mean; Correlation coefficient: spearman.
GPX7-associated microRNAs
To predict GPX7-targeting mirRNAs we used (i) cancer regulome (http://explorer.cancerregulome.org/) (correlation of Abs = 0.4; Max results: 200; p < 0.05 ); (ii) DIANA-miRPath v.3 (http://snf-515788.vm.okeanos.grnet.gr/)51; (iii) Targetscan (http://www.targetscan.org/)52; (iv) miRWalk 3.0 database (http://mirwalk.umm.uni-heidelberg.de/)53,54; and (iv) miRDB database (http://mirdb.org/)55,56,57. All candidate miRNAs were compared with all databases. To understand the signaling pathways associated the miRNA signatures, we performed the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis58,59,60 in DIANA-mirPath v.3. p < 0.05 and false discovery rate (FDR) were considered to estimate statistical significance in enrichment analysis.
Tumor immune estimation resource (TIMER) analysis
The TIMER database (http://timer.comp-genomics.org/)61,62 was used to estimate the correlations between GPX7 expression and the abundance of immune cell infiltration levels. It uses a deconvolution method to deduce the abundance of tumor-infiltrating immune cells based on gene expression profiles. In this study, we adopted CIBERSORT, CIBERSORT-ABS, EPIC, quanTIseq, TIMER, xCell and MCPcounter immune deconvolution algorithms to cover as many immune cell types as possible. Tumor purity was considered when calculating Spearman’s correlation, and a p-value <0.05 was considered statistically significant.
TISIDB database analysis
TISIDB is an integrated repository web portal to analyze interactions between tumors and the immune system63. It integrates multiple types of data resources in oncoimmunology, including literature mining results from the PubMed database and TCGA. The TISIDB was used to assess the role of GPX7 in the tumor–immune interplay.
Statistical analysis
R software (v4.0.5) was employed to implement the statistical analyses of the study. p values < 0.05 were considered significant unless otherwise specified. The relationships of GPX7 expression and clinicopathological features were estimated using the Chi-Squared Test. Analyses with more than two groups were performed by one-way ANOVA test. Statistical significance was denoted by *p < 0.05, **p < 0.01, ***p < 0.001. The associations of the gene expression levels were analyzed using a non-parametric Spearman’s rho test. The strength of the correlation was determined using the following: (r) ≥ 0.7 indicated a strong correlation; r < 0.7 and r ≥ 0.3 indicated a moderate correlation, and r < 0.3 indicated a weak correlation.
Results
GPX7 is overexpressed in gliomas tissues and cell lines
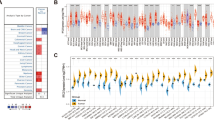
Changes in expression levels of most glutathione peroxidases have been reported in several tumors. However, to the best of our knowledge, few studies have investigated the GPX7 expression and its impact on most cancers. To this end, we explored the expression patterns of GPX7 from a pan-cancer perspective, using microarray data through the differential analysis tool of the Oncomine database. Strikingly, we observed that GPX7 was overexpressed in 19 datasets (brain, breast, esophageal, gastric, liver cancers, leukemia, melanoma, myeloma and sarcoma) and underexpressed in lymphomas (Fig. 1a), indicating that the dysregulation of this glutathione peroxidase was a common phenomenon across several tumors.

Transcription levels of GPX7 in different types of human cancers compared with normal tissues (Oncomine database). (a) The number of datasets with statistically significant mRNA upregulated (red) or downregulated expression (blue) of GPX7. The numbers in the colored cell represent datasets meeting the threshold (see “Material and methods”). The gene rank was analyzed by the percentile of the target gene at the top of all genes measured in each research. The cell color is determined by the best gene rank percentile for the analyses within the cell. Significant Unique Analyses indicate that the queried gene is significantly different in studies. Total Unique Analysis indicates the total number of studies that contain the queried gene. (b) GPX7 expression levels in distinct glioma cohorts. Rembrandt microarray cohort (Normal samples = 28 samples; LGG samples = 225 samples; GBM samples = 219 samples); Gravendeel microarray cohort (Normal samples = 8 samples; LGG = 117 samples; GBM = 159 samples); Kamoun microarray cohort (Normal samples = 9 samples; LGG = 154 samples; GBM = 16 samples); Chinese Glioma Genome Atlas (CGGA) RNA-seq cohort (Normal samples = 20 samples; LGG = 625 samples; GBM = 388 samples); TCGA RNA-seq cohort (LGG = 518 samples; GBM = 163 samples). Red boxplots: Tumor samples from TCGA. Grey boxplots: Normal samples from GTEx (Genotype–Tissue Expression database) (N = 207 samples). (c) mRNA expression of GPX7 in different types of cell lines of human cancer from CCLE (Cancer cell line encyclopedia). The red arrow indicates the glioma cell lines (66 cell lines). The number next to the lineage name represents the number of cell lines in the lineage. The expression ranks in gliomas using Affymetrix GeneChip data. Statistical significance is denoted by *p < 0.05, **p < 0.01, ***p < 0.001. (d) GPX7 gene expression profiling during the cell cycle in T98G cells (Human Glioblastoma Cell line) (GDS3364 from GSE8537 dataset)35.
Subsequently, we focused our analysis on the differential expression of GPX7 in all brain cancer datasets deposited on Oncomine. As presented in Table 1, GPX7 was found to be higher in gliomas, especially in astrocytic, oligodendroglial and mixed gliomas. To further expand and validate these results, we analyzed five transcriptome datasets: Rembrandt microarray27, Gravendeel microarray28, Kamoun microarray29, CGGA RNA-seq30, and TCGA RNA-seq. As shown in Fig. 1b, we consistently showed that GPX7 was overexpressed in gliomas compared to normal samples in all datasets.
By assembling the Cancer Cell Line Encyclopedia (CCLE), we further explored whether the overexpression pattern of GPX7 detected in LGG and GBM tissues would be similar in glioma cell lines. Accordingly, the vast majority of glioma cell lines highly expressed GPX7 (Fig. 1c) and this overexpression was cell cycle-dependent manner (Fig. 1d).
Together, these data support that GPX7 overexpression may be involved in glioma tumorigenesis, which appears to be differentially regulated by molecular mechanisms during the progression of the cell cycle, perhaps being modulated by various factors of the tumor microenvironment, especially in response to exposure to oxidative stress (ROS accumulation). These data prompted us to further investigate the relationship between the expression of this gene with the clinical and molecular parameters in LGG and GBM.
Clinicopathological and molecular features of adult LGG and GBM strongly impacts GPX7 expression
Leveraging the TCGA and CGGA datasets, we next explored the correlation between GPX7 expression and the clinicopathological/molecular parameters of gliomas (WHO grade 2–4). Notably, in both cohorts, GPX7 was prominently higher in GBM than in LGG tumors (Oligoastrocytoma; Oligodendroglioma; Astrocytoma), while the expression of GPX7 was significantly higher as pathological grade increased (Fig. 2a–d). Besides, GPX7 expression was found to be significantly correlated with additional pharmaceutical therapy (p = 0.0054), primary therapy outcome success (p = 0.000038), new tumor event after initial treatment (p = 0.0000029), Postoperative rx tx (p = 0.038), radiation therapy (p = 0.0000000013), sensory changes (p = 0.0027) and visual changes (p = 0.01) (Supplementary Table S1) in TCGA cohort. Furthermore, in the CGGA dataset, the elevated expression of GPX7 was correlated with radiochemotherapy status and progression status, consistent with the findings seen in the TCGA dataset (Supplementary Table S2). In view of the aforementioned findings, GPX7 is abundantly expressed in aggressive gliomas, suggesting that GPX7 might be involved in the malignant progression of gliomas.

GPX7 is highly expressed in gliomas and is significantly associated with the advanced stage of tumors. (a) Relative mRNA expression of GPX7 in histopathological types of gliomas; (b) Relative mRNA expression of GPX7 in different gliomas WHO grades (2, 3 and 4) from the TCGA cohort; (c) Relative mRNA expression of GPX7 in histopathological types of gliomas from the CGGA cohort; (d) Relative mRNA expression of GPX7 in different gliomas WHO grades (2, 3 and 4) from the CGGA cohort. Statistical significance was denoted by asterisks: *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001; ****p ≤ 0.0001.
Gliomas, like other tumors, show high genetic heterogeneity69,70,71,72. Numerous studies revealed that the most common somatic chromosomal changes in these tumors are complete or partial loss of chromosomes 1, 7, 10 and 1973,74,75,76,77,78,79,80. Intriguingly, our data showed that higher GPX7 expression was significantly correlated with 1p/19q non-codeleted group, carrying concomitant gain of chr7/loss of chr10, mostly seen in GBM patients (Fig. 3a; Supplementary Fig. S1). These findings led us to hypothesize that the molecular classification proposed for LGG and GBM could also impact GPX7 expression.

GPX7 expression was correlated with CNAs (Copy Number Alterations), IDH1 mutational status and 1p/19q co-deletion. (a) Heatmap integrating GPX7 expression (Log2—Norm Count + 1), CNAs (Copy Number Alterations) status (Log2—Tumor/Normal) of chromosomes 1, 7, 10 and 19, histology and WHO grade of LGG and GBM cohorts from TCGA. GPX7 mRNA expression in molecular subtypes of (b) Low-Grade Gliomas (LGG) cohort, according to WHO81 (IDH-mutant, 1p/19q codeleted; IDH-mutant, 1p/19q non-codeleted; and IDH-wildtype, 1p/19q non-codeleted group) and of (c) GBM cohort, according to Verhaak82 (Proneural; Classical; Mesenchymal; Neural) from TCGA database. (d) GPX7 expression correlates with IDH-1 status in CGGA gliomas. (e) GPX7 expression correlates with 1p/19q co-deletion in CGGA gliomas. *p < 0.05, **p < 0.01, ***p < 0.001.
The most recent guidelines for the classification of gliomas recommend that LGG should be categorized into three major groups81: (1) IDH-mutated 1p/19q codeleted group; (2) IDH-mutated 1p/19q non-codeleted group (most of them have mutations in TP53 and ATRX); (3) IDH-wildtype 1p/19q non-codeleted group. Here, using this classification, we showed that GPX7 was significantly higher in the TCGA-LGG patients with wild-type IDH1, TP53 and ATRX, without 1p/19q non-codeletion (Fig. 3b). Subsequently, using the TCGA-GBM cohort, we considered the molecular subtypes proposed by Verhaak (Proneural; Classical; Mesenchymal; Neural)82 as well as the IDH1 status, MGMT methylation status, and G-CIMP (Glioma CpG island methylator phenotype) status. As noted in Supplementary Fig. S2, GPX7 expression varied considerably depending on the CIMP status and IDH1 status. Remarkably, GPX7 was increased in wildtype IDH1 non-GCIMP neural GBMs (Fig. 3c). To validate our results, CGGA samples were subsequently used to verify these relationships. GPX7 expression was also significantly associated with IDH mutation status (Fig. 3d) and 1p/19q co-deletion status (Fig. 3e).
Lastly, concerning the most commonly mutated genes in LGG, wild-type TERT, IDH1/2, CIC, NOTCH1, PIK3CA and FUBP1 were significantly enriched in TCGA-LGG patients with high expression of GPX7 (Supplementary Fig. S3a–g). Additionally, patients harboring TP53, ATRX, NF1, PTEN and EGFR mutations also over-expressed GPX7 (Supplementary Fig. S3h–l).
In summary, our results indicate that the overall GPX7 expression varied significantly as a function of histopathological grade and is highly associated with glioma progression. Also, GPX7 expression might be correlated to chromosomal changes, mainly concomitant gain of chr7/loss of chr10 (primarily seen in GBM), as well as mutations in TP53, ATRX, NF1, PTEN and EGFR (mostly observed in LGG).
The overexpression of GPX7 correlates with unfavorable prognosis in adult LGG tumors
Next, we applied Kaplan–Meier survival curves to assess the GPX7 prognostic role in TCGA LGG and GBM cohorts. The results illustrated that higher expression of GPX7 was markedly correlated with worse overall survival (OS) (HR = 2.8; Logrank p = 4.4e–08) and disease-free survival (DFS) (HR = 1.9, Logrank p = 3.6e–05) in LGG (Fig. 4a). Furthermore, considering all histological subgroups of LGG, higher GPX7 was significantly correlated with poor OS in astrocytomas (HR = 2.6; Logrank p = 0.017) (Fig. 4b), oligoastrocytomas (HR = 5.8; Logrank p = 0.0085) (Fig. 4c) and oligodendrogliomas (HR = 3.1; Logrank p = 0.013) (Fig. 4d) and poor DFS in astrocytomas (HR = 2.2; Logrank p = 0.029) (Fig. 4b). However, no significant correlation was observed between the expression of GPX7 and the prognosis of GBM (Fig. 4e). Thus, we naturally extended our analysis to other glioma datasets, and in all of them, we consistently found that overexpression of GPX7 was associated with a worse prognosis (Supplementary Fig. S4–S5). In conclusion, these data suggested that GPX7 had a significant effect on the prognosis of LGG.

Kaplan–Meier survival curves are based on high (red) and low (blue) expression of GPX7 in LGG and GBM from the TCGA database. Overall survival (OS) (left) and disease-free survival (DFS) (right) of (a) All histological subgroups of LGG (n = 514), (b) Astrocytomas (n = 95), (c) Oligoastrocytomas (n = 76), (d) Oligodendrogliomas (n = 114) and (e) Glioblastomas (n = 162). X-axis: patients’ survival duration (months); Y-axis: patients’ survival rate. p-Values for all survival analyses have been calculated using the log-rank test.
Hypomethylation of GPX7 promotes its expression in LGG tumors
DNA methylation is one of the best-characterized epigenetic modifications generally occurring on cytosines that precede a guanine, which are concentrated in large clusters throughout the genome, called CpG islands. Consequently, DNA methylation can impact DNA conformation, chromatin structure, DNA–protein interactions, DNA stability and can modify gene expression. Furthermore, both initiation and late stages of oncogenesis may be led by DNA methylation changes, given that epigenetic changes have been demonstrated in multiple cancers, including gliomas83,84. In this regard, we next sought to uncover the molecular mechanisms that contribute to the high expression of GPX7 in gliomas. For this, we analyzed the methylation levels of all CpG islands based on the regions defined by the UCSC Genome Browser (First Exon, 3’UTR, Body, TSS1500 and TSS200), S-Shore (2000 bp region 3′ adjacent to CpG island), S-Shelf (2000 bp region 3′ adjacent to S-Shore), and N-Shore (2000 bp region 5′ adjacent to CpG island), of LGG and GBM cohorts from TCGA database. Overall, both cohorts exhibited extensive hypomethylation levels at cg02453146, cg23272399, cg22129364, cg16557944, cg20950465, cg11953272 and cg26251270 CpG sites and higher hypermethylation levels at cg00998379 CpG site (Fig. 5a, b; Supplementary Table S3). Curiously, four regions (cg02453146, cg23272399, cg09161043 and cg18087326) from LGG stood out forming clusters with distinct methylation patterns, which might reflect the complexity/heterogeneity of the methylation program involved in these tumors.

Dynamics of the DNA methylation across CpG sites of GPX7 in LGG and GBM tumors. (a) Heat-map showing the methylation levels of GPX7 among different CpGs sites (probes) integrating ethnicity, race, age, vital status and genomic regions of CpG sites from LGG; (b) Heat-map showing the methylation levels of GPX7 among different CpGs sites integrating ethnicity, race, age, vital status and genomic regions of CpG sites from GBM. (c) Pearson correlation between the degree of methylation (β-value) of each CpG site of GPX7 and its expression level (Log2 – TPM + 1) in LGG; Aggregation method: mean; (d) Pearson correlation between the degree of methylation (β-value) of each CpG site of GPX7 and its expression level (Log2 – TPM + 1) in GBM. Aggregation method: mean.
Considering the aforementioned results, we sought to ascertain whether hypomethylation was the cause of increased GPX7 expression. We assessed the levels of GPX7 mRNA and correlated them with the methylation status of each CpG site (Fig. 5c, d). Strikingly, the methylation levels for all CpG sites revealed a significant negative correlation with mRNA levels in LGG tumors (Aggregation ρ = − 0.48, p < 2.2e−16) (Fig. 5c). To further strengthen these results, analyzing an independent dataset from GEO database, we verified that short-term exposure (96 h) of cultured glioblastoma cell line (GLI56) to 5-aza-2′-deoxycytidine (5-aza-dC) [5 μM], a global DNA demethylating agent, induced a substantial increase in the GPX7 expression (Supplementary Fig. S6), thus strongly suggesting that upregulation of GPX7 is tightly regulated by DNA methylation.
Since we confirmed the correlation between methylation and regulation of GPX7 mRNA expression, we next analyzed the impact of the methylation pattern of each CpG site on overall survival (OS) in LGG patients. Consistently, seven CpG sites (cg02453146; cg23272399; cg11953272; cg03330022; cg09161043) distributed in different genomic regions (Body; TSS200; TSS1500) were significantly correlated with the prognosis of LGG (Supplementary Table S3). This evidence suggests that upregulation of GPX7 was caused by DNA hypomethylation, with an impact on poor OS in patients with LGG.
GPX7 overexpression is highly correlated with histone acetyltransferase 1 (HAT1) expression and with H3K9ac and H3K27ac marks
It is well known that the crosstalk between both DNA hypomethylation and histone acetylation is involved in the upregulation of gene transcription85,86,87. In particular, histone acetylation, which is catalyzed by histone acetyltransferases (HATs), disrupts the electrostatic interaction between histones and DNA, thus conferring positive effects on gene expression. Based on cell localization, HATs are subdivided into (i) Type A HATs (mainly expressed in the nucleus) and (ii) Type B HATs (mainly expressed in the cytoplasm)87,88. Therefore, we speculate that GPX7 expression could be correlated with HATs and acetylation marks in LGG and GBM.
Initially, we observed that most HATs were differentially expressed in LGGs and correlations between HATs and GPX7 expression were mostly seen in LGG (Supplementary Fig. S7 and Supplementary Table S4). Notably, in both cohorts, the highest positive correlation was with histone acetyltransferase 1 (HAT1) (LGG: ρ = 0.71, p = 1.00e–51; GBM: ρ = 0.31; p < 5.50e–05) and among the LGG, this correlation was significantly higher in anaplastic oligodendrogliomas (WHO grade 3) (ρ = 0.64, p = 1.7e−07) and in mixed glioma (WHO grade 3) (ρ = 0.62, p = 1.9e−05) (Fig. 6a), suggesting that GPX7 might be epigenetically regulated by HAT1-mediated acetylation, especially in high-grades.

H3K9ac and H3K27ac marks can alter GPX7 expression. (a) Correlation between GPX7 and HAT1 expression according to WHO grades (2 and 3) and histology of TCGA-LGG tumors. NOS: Not Otherwise Specified. (b) Representative UCSC genome browser view of GPX7 genomic region showing chromatin immunoprecipitation (ChIP) enrichment for H3K9ac and H3K27ac (active chromatin marks) using ChIP-seq data from LN229 (GBM cell line) (GSE109340), GBM12-5199 (PDX model of GBM)89, GBM12-3080 (PDX model of GBM)89, GBM12-5199 (PDX model of GBM)89, GBM12-3080 (PDX model of GBM)89, Brain tumor initiating cells (BTICs)90, differentiated glioma cells (DGCs)90. DNase clusters: shows chromatin accessibility following binding of trans-acting factors in place of a canonical nucleosome. The display for this track shows site location and signal value as grayscale-colored items where higher signal values correspond to darker-colored blocks. This track is a composite annotation track containing multiple subtracks, one for each cell type. Layered H3K27Ac: This track shows the enrichment levels of the H3K27Ac histone mark across the genome as determined by a ChIP-seq assay. Also, this mark is thought to enhance transcription, possibly by blocking the spread of the repressive histone mark H3K27Me3. GPX7 expression (TCGA): shows RNA expression level for each TCGA tumor (LGG and GBM) in GENCODE canonical genes. The gene scores are a total of all transcripts in that gene. TF Clusters: This track shows transcription factor (TF) binding sites derived from a large collection of ChIP-seq experiments. A gray box encloses each peak cluster of transcription factor occupancy. The darkness of the box is proportional to the maximum signal strength observed in any cell type contributing to the cluster. ENCODE cCRES track: This track displays the ENCODE Registry of candidate cis-Regulatory Elements (cCREs) in the mouse genome. cCREs are the subset of representative DNase hypersensitive sites across ENCODE samples supported by either histone modifications (H3K4me3 and H3K27ac) or CTCF-binding data. CCREs are colored and labeled according to classification by regulatory signature: Red (promoter-like signature); orange (proximal enhancer-like signature); yellow (distal enhancer-like signature); pink (DNase-H3K4me3); blue (CTCF-only).
Next, to directly show a functional link between the expression of GPX7 and the acetylation of histones, we analyzed the distributions of active chromatin marks, marked by histone 3 lysine 9 and/or lysine 27 acetylation (H3K9ac or H3K27ac, respectively), mapped by ChIP-Seq using data from the ENCODE Project in UCSC Genome Browser database in different human glioma models. Strikingly, H3K9ac and H3K27ac marks were predominantly enriched near of active regulatory elements and in the TSS region of the GPX7 in all models, as indicated by the colocalization of DNase clusters (DNaseI hypersensitivity), transcription factors (TF clusters) peaks and ENCODE cCREs peaks, which also correlates with an increase in GPX7 expression (Fig. 6b), suggesting that changes in these chromatin marks could potentially affect the modulation of GPX7 expression.
miRNAs potentially regulate GPX7 expression in LGG and GBM
Next, to verify whether miRNAs could regulate GPX7 expression, we identified miRNAs positively or negatively correlated with GPX7 in LGG and GBM datasets from the TCGA (Fig. 7a, b and Supplementary Tables S5–S6). Upon identifying these miRNAs signatures in both cohorts, we focused on the gene-miRNA interactions with a primary focus on predicting the miRNAs that potentially interact with GPX7. A total of 8 miRNAs (hsa-mir-29c-3p; hsa-mir-137-3p; hsa-mir-767-5p; hsa-miR-196a-5p; hsa-miR-196b-5p; hsa-miR-92b-5p, hsa-miR-885-3p and hsa-miR-139-3p) were identified for LGG and 3 miRNAs (hsa-miR-29c-3p, hsa-let-7e-5p, hsa-miR-29b-3p) were identified for GBM.

Genome-wide associations among GPX7 and miRNAs in the LGG and GBM. (a) Circos plots of predicted miRNAs signatures co-expressed with GPX7 in LGG and GBM patients from the TCGA database. Only genes with a correlation of Abs = 0.4 and p < 0.05 are shown in the circus plots. Chromosomes 1–Y are shown outer in the right half of the circle. miRNA genes are represented by purple lines near their chromosomal locations. The outer ring displays cytogenetic bands. In this case, lines connecting two dots represent the statistically significant correlation between two selected features: miRNA expression and GPX7 expression. Heat-maps of significant KEGG pathways of GPX7-associated miRNAs by utilizing the DIANA-miRPath in (b) TCGA-LGG and (c) TCGA-GBM. Each row represents a miRNA, and each column represents the KEGG pathway51,58,59,60. Gradient from red to yellow indicates a higher to lower p-value (Log).
To dissect the biological significance of the above selected miRNAs, DIANA-miRPath v3.0 (TargetScan database) was used to identify the signaling pathways in which these miRNAs may be involved. As shown in Fig. 7b, in LGG signature, four miRNAs (hsa-mir-29c-3p; hsa-mir-767-5p; hsa-miR-196a-5p and hsa-miR-196b-5p) prominently modulated ECM-receptor interaction, while two miRNAs (hsa-mir-29c-3p and hsa-miR-885-3p) importantly contributed to glioma development. Likewise, the miRNAs signature from GBM also affected the ECM-receptor interaction pathway (Fig. 7c). Furthermore, we also noticed a sharp correlation between all miRNAs and fatty acid biosynthesis in these tumors. These results showed that GPX7-associated miRNAs dysregulated different pathways in gliomas, converging mainly to the regulation of the extracellular matrix, which has a dramatic influence on glioma invasiveness and aggressiveness91.
GPX7 could be specifically upregulated in glioblastoma models treated with different therapies
Malignant primary brain tumors are a leading cause of cancer mortality in children and young adults, with few therapeutic options92. To date, the standard treatment for high-grade gliomas (HGGs) consists of surgical tumor resection followed by fractionated radiotherapy and chemotherapy with alkylating agents, such as temozolomide (TMZ)93,94,95, or with molecular targeted drugs (for recurrent tumors), such as bevacizumab (BEV) (an antiangiogenic drug)96,97,98,99 and irinotecan (a DNA topoisomerase I inhibitor)97,100. Furthermore, the consensus in cancer research suggests that most chemotherapeutics elevate intracellular levels of reactive oxygen species (ROS)101, resulting in oxidative injury and cell damage.
In light of this, we proposed that different therapeutic strategies available for gliomas might affect GPX7 expression, considering that GPX7 acts to reduce oxidative stress8. To test this hypothesis, we then delved into the therapy-related microarray datasets available from the GEO database for GBM. From the GSE43452 dataset36, we found that the GPX7 was significantly increased in the U87 cell line after treatment with 20 µM of TMZ for 24 h (p = 0.0106) (Fig. 8a). Consistent with this, we also observed a similar increase in U87-EV human GBM xenograft tumor treated with bevacizumab (10 mg/kg of body weight) when compared with non-treated tumors (GSE39223 dataset)37 (Fig. 8b).

GPX7 expression in glioma models treated with chemotherapy. (a) The relative GPX7 mRNA expression of human glioblastoma cell line (U87MG) treated with temozolomide at 20 µM for 24 h (GDS4808 from GSE43452 dataset)36. Two-tailed Wilcoxon rank-sum test was used (p-value = 0.0106); (b) Relative GPX7 mRNA expression in U87MG-EV human glioblastoma xenograft tumor treated with bevacizumab (10 mg/kg body weight) (GDS5672 from GSE39223 dataset)37. Results are shown as mean ± SD. Statistical significance is denoted by ***p < 0.001 compared with control. **p < 0.01 compared with control. *p < 0.05 compared with control; (c) Box plot (left) depicting the expression of GPX7 (probe 213170_at) in GBM patients annotated with chemotherapy responses according to the Response Evaluation Criteria in Solid Tumors (RECIST) criteria. Graphs show normalized gene expression in irinotecan non-responders and responders patients. ROC plot of non-responders (N = 10) and responders (N = 23) of GBM patients treated with Irinotecan based on overall survival (OS) at 16 months. Error bar ± SD. AUC: area under the curve. PPV, positive predictive value; TPR: true positive rate.
Next, in order to explore the predictive potential of GPX7 expression as a chemotherapy response predictor, we performed ROC (Receiver Operating Characteristic) in an independent GBM dataset (n = 454)45 treated with multiple chemotherapeutic agents (Supplementary Fig. S8), as described in the methods. For irinotecan treatment, the non-responder group exhibited higher expression of GPX7 than the responder group (Fold-change = 1.4; p = 0.03) (Fig. 8c). Similarly, the ROC curve showed that increased GPX7 expression could perfectly distinguish irinotecan therapy respondents from non-responders (AUC = 0.726; p = 2.2e–02) (Fig. 8c). Together, these findings support the idea that changes in GPX7 expression levels are likely related to the induction of oxidative stress and ROS-mediated cell injury induced by distinct drugs approved to treat gliomas. Also, our dataset supports the application of GPX7 expression to stratify and identify GBM patients that are likely to benefit from irinotecan treatment, given that enhanced GPX7 expression may contribute to therapeutic resistance in GBM subpopulations.
The biological function of co-expressed genes related to GPX7 in LGG and GBM
To better understand the underlying molecular mechanisms by which GPX7 modulates the biological processes in gliomas, we screened out the GPX7 co-expressed gene signatures by the LinkFinder module in the LinkedOmics. In total, 4555 genes were related to GPX7 expression (1716 positively correlated and 2838 negatively related) in LGG, whereas in GBM, we found 3863 GPX7-related genes (2648 positively correlated and 1215 negatively related) (Fig. 9a). Using gene set enrichment analysis (GSEA), we observed that the GO biological process profiles were markedly different in both cohorts. In LGG, GO terms mainly fit into immune mechanisms involving both innate and adaptive immunity, type I interferon production and regulation of synaptic transmission (Fig. 9b). By contrast, marked upregulation of the metabolic regulation of mitochondrial dynamics, translation and ribosomal synthesis signature along with downregulation of a synaptic transmission signature was seen in GBM (Fig. 9c). Our data here provide a basis to explore clinically relevant biological processes involved in gliomagenesis that can be used as potential targets in translational medicine.

Functional enrichment analysis of GPX7-related genes in LGG and GBM cohorts from. TCGA. (a) Venn diagram of genes that positively (Left) and negatively (Right) correlated with GPX7 expression, showing shared genes in LGG and GBM. (b) Functional annotation using GSEA shows an overrepresentation of Gene Ontology (GO) terms of upregulated (blue) and downregulated (orange) genes co-expressed with GPX7 in LGG. (c) Functional annotation using GSEA shows an overrepresentation of Gene Ontology (GO) terms of upregulated (blue) and downregulated (orange) genes co-expressed with GPX7 in GBM. Dark blue and orange: FDR ⩽ 0.05; Light blue and orange: FDR > 0.05. Abbreviations: FDR: false discovery rate; GO: Gene ontology.
GPX7 expression is associated with immune cell infiltration
Given the heterogeneity of the tumor immune microenvironment, the close relationship between immunological features and the therapeutic response in gliomas102,103, and the association found between GPX7-co-expressed genes with immune-related gene ontology terms, especially in LGG, we reasoned that GPX7 expression could have potential to impact the immune cell infiltration. Here, we found that GPX7 was negatively associated with the tumor purity in LGG (ρ = − 0.486; p = 8.63e−30) (Fig. 10 and Supplementary Table S7) but positively correlated to it in GBM (ρ = 0.311; p = 2.02e−04) (Fig. 11 and Supplementary Table S7). These results are in accordance with the finding of gene ontology terms for GPX7-co-expressed genes being related to stromal reactions (e.g.: immune cell infiltration and IFN-I response) in LGG, but being mainly related to metabolic processes in GBM, suggesting that while in LGG GPX7 activation directly impacts the tumor microenvironment, in GBM the affected processes are tumor-cell intrinsic.

Correlation of GPX7 expression with immune infiltration level in LGG (TCGA dataset) via Tumor Immune Estimation Resource (TIMER) database. All spearman correlations were adjusted for tumor purity.

Correlation of GPX7 expression with immune infiltration level in GBM (TCGA dataset) via Tumor Immune Estimation Resource (TIMER) database. All spearman correlations were adjusted for tumor purity.
Moreover, in LGG, GPX7 expression was positively correlated with multiple immune-stromal cells, such as B cells (ρ = 0.189; p = 3.18e−05), common lymphoid progenitors (ρ = 0.387; p = 1.69e−18), monocytes (ρ = 0.382; p = 4.70e−18), M1 (ρ = 0.148, p = 0.001) and M2 macrophages (ρ = 0.354, p = 1.42e−15), mast cells (ρ = 0.319; p = 8.87e−13), myeloid dendritic cells (ρ = 0.572; p = 6.20e−43), neutrophils (ρ = 0.594; p = 4.65e−47), CD4 + T cells (LGG, ρ = 0.503; p = 4.77e−32), Treg cells (ρ = 0.180; p = 7.39e−05), CD8+ T cells (ρ = 0.162; p = 0.0003), NK cells (ρ = 0.198; p = 1.29e−05) and cancer-associated fibroblasts (CAFs, ρ = 0.319; p = 8.92e−13) (Fig. 10; Supplementary Table S7). Furthermore, after purity-related adjustment, we found that GPX7 expression was related to high intratumor expression of most markers of T cells (general) (CD3E, CD3D, CD2), B cells (CD19, CD79A), monocytes (CD86, CD115), TAMs (CCL2, CD68, IL10), M2 Macrophages (CD163, VSIG4, MS4A4A), neutrophils (CD11b, CCR7), dendritic cells (HLA-DPB1, HLA-DQB1, HLA-DRA, HLA-DPA1, BDCA-1, BDCA-4), CD4+ T cells (CD4, CD45RA), Th1 cells (T-bet, STAT4, STAT1, IFN-g, TNF-a), Th2 cells (GATA3, STAT6, STAT5A) and Treg cells (FOXP3, CCR8, STAT5B, TGFβ) (Supplementary Table S8). Thus, these data indicated that high GPX7 expression is associated with a highly inflammatory microenvironment and immune exhaustion in these tumors.
Otherwise, in GBM, GPX7 only positively correlated with common lymphoid progenitors (ρ = 0.485; p = 1.89e−09), CD8+ (ρ = 0.290; p = 0.0005), myeloid-derived suppressor cells (ρ = 0.379; p = 4.93e−06) and CAFs (ρ = 0.220; p = 0.009), while it was negatively correlated with B cells (ρ = − 0.322; p = 0.0001), eosinophils (ρ = − 0.413; p = 5.13e−07), monocytes (ρ = − 0.348; p = 3.03e−05), CD4 + T cells (ρ = − 0.354; p = 2.08e−05), NK cells (ρ = − 0.393; p = 2.02e−06) and NKT cells (ρ = − 0.331; p = 0.0006) (Fig. 11 and Supplementary Table S7). Besides, exploring the correlation between GPX7 and sets of immunological markers using the TIMER2.0 database, we also observed significant correlations between GPX7 and the monocyte marker CD115, the M1 macrophage marker IRF5, the neutrophil marker CD11b, the NK marker KIR2DL3, CD4+ marker CD11c, the Th2 marker STAT6, the Tfh marker BCL6, the Th17 marker STAT3 and Treg markers FOXP3, STAT5B, TGFβ (Supplementary Table S8), therefore suggesting that GPX7 expression differently affect tumor immune-stroma in these types of cancer.
Consistently, closer inspection of immune-related signatures from the TISIDB database revealed that a variety of subtypes of tumor-infiltrating lymphocytes (TILs) were significantly affected by GPX7 expression in LGG including γδ T cells, conventional CD4+ T cells (memory CD4+ T cells—Tcm CD4), CD8+ T cells (Activated and Central memory CD8+ T cells), B cells (memory and immature B cells), NK cells, NKT cells, myeloid-derived suppressor cell (MDSC), activated dendritic cell, and mast cell. Differently, in GBM, GPX7 only showed a close connection with γδ T cells, activated CD4+ T cells, and activated and immature B cells (Fig. 12). Based on these results, it is conceivable that the correlation of immune cells and GPX7 expression may be characteristic of tumor/tissue type, which emphasizes the uniqueness of immune infiltration in gliomas.

Correlation between GPX7 expression and tumor-infiltrating immune cells. Left panel: Correlative heat-map between GPX7 and 28 tumor-associated immune cells (Vertical axis) in a pan-cancer cohort (Horizontal axis). The colors indicate the correlation coefficients; Upper panel: correlation between GPX7 expression and immune-related signatures from (i) gamma delta T cells (γδ T cells); (ii) central memory CD4+ T cells (Tcm CD4+ T cells); (iii) activated CD8+ T cells (Act CD8+ T cells); (iv) central memory CD8+ T cells (Tcm CD8+ T cells); (v) NK cells; (vi) NKT cells; (vii) memory B cells; (viii) immature B cells; (ix) Myeloid-derived suppressor cells (MDSC cells); (x) activated dendritic cells; (xi) mast cells in low-grade gliomas (LGG). Lower panel: correlation between GPX7 expression and immune-related signatures from (i) gamma delta T cells (γδ T cells); (ii) activated CD4+ T cells (Act CD4+ T cells); (iii) activated B cells; (iv) immature B cells in glioblastoma (GBM).
The burgeoning field of immune cell migration into solid tumors has created new and exciting opportunities in translational cancer immunotherapy104. In this context, it is now clear that the migration of both effector and suppressive immune cell types to the tumor microenvironment (TME) is controlled by a plethora of chemokines. As we found immune cells to be potentially influenced by GPX7, we next hypothesized that GPX7 could be acting to modulate immune cell migration. To test this, we compared GXP7 expression with key chemokines and their corresponding receptors. As shown in Supplementary Table S9, the most relevant chemokines correlated with GPX7 were: (i) in LGG, CCL1/2/5/20/22/25 and CXCL9/10/11/16; (ii) and in GBM, CCL4/16 and CXCL1. Concerning the chemokine receptors, CCR1/2/5, CXCR2/3/4/6, XCR1 and CX3CR1 showed the greatest correlation with GPX7 in LGG, while CCR1/5, CXCR2 and CX3R1 were the most significantly correlated in GBM.
To gain a better understanding of the modulation of TILs migration into the LGG TME, further associations between GPX7 and immunomodulators, including immune checkpoint receptors, activating receptors and MHC molecules, were examined. We identified striking positive correlations with most immune checkpoints and immune suppressive receptors in LGG, such as TIM3 (HAVCR2), IL10RB (interleukin 10 receptor subunit beta), LGALS9 (galectin 9), PDCD1LG2 (programmed cell death 1 ligand 2), TGFB1 (transforming growth factor beta 1) and TGFBR1 (transforming growth factor beta receptor 1). In contrast, only IL10RB was highly associated in GBM (Supplementary Fig. S9). Amongst the immunostimulators, CD276, CD40, CD48, CD86, IL6, MICB, TMEM173 and TNFRSF8 presented the highest correlation coefficients in LGG, whereas TNFRSF25, IL6R displayed the highest correlation coefficients in GBM (Supplementary Fig. S10). Moreover, almost all MHC‐related genes were highly correlated with GPX7 expression in LGG, but weakly correlated in GBM (Supplementary Fig. S11).
Finally, we investigated the relationship between each immune cell type and overall survival (OS) in LGG and GBM patients. All patients were initially divided into high and low-expressing groups according to the TILs and GPX7 expression. Interestingly, the survival curves showed that low expression of GPX7 along with low immune infiltration of CD4+ T cells, neutrophil, myeloid dendritic cell (mDCS) and cancer associated fibroblast (CAF) have better prognosis than the high TIL- and GPX7-expressing group in LGG (Fig. 13). In contrast, no association with survival was observed in GBM.

Effect of GPX7 expression and immune infiltration on TCGA-LGG patients overall survival (OS). Kaplan–Meier plot displaying differences in OS among patients stratified by both the predicted infiltration level of immune cells (CD4+ T cells, neutrophil, myeloid dendritic cell and cancer-associated fibroblast) and GPX7 expression level in LGG. High and low immune infiltration cutoffs were determined based on the infiltration score greater than or less than 0, while high and low GPX7 expression cutoffs were defined using the median. The prognostic signature built by the immune infiltration of CD4+ T cells, neutrophil, myeloid dendritic cell and GPX7 expression was profiled by TIMER algorithm, while the signature of cancer associated fibroblast and GPX7 expression was profiled by TIDE. P-values were calculated using the log-rank test and vertical hash marks indicate censored data.
Together, these findings strongly suggest that GPX7 may be involved in tumor inflammatory response, which might be responsible for modulating the immune molecules and TILs migration, impacting patient outcomes, especially those with LGG.
Discussion
Gliomas are characterized by a complex TME which plays a critical role in tumor invasiveness, malignancy and therapy failure. Current evidence suggests that the reactive oxygen species (ROS) derived from redox (reduction–oxidation) imbalance in the TME may contribute to genomic damage, promoting DNA mutations and activating oncogenes105, and also affect intracellular signal transduction of multiple cellular pathways, thus conferring specific conditions for initiation and malignant progression of many human tumors, including gliomas105,106,107,108,109. As part of the antioxidant system in mammals, the GPXs family is a major regulator of cellular redox state7,8. In particular, the non-selenocysteine-containing GPX7 has been shown to be essential for the maintenance of redox homeostasis through oxidative protein folding5,6,7 and releasing of the non-targeting short interfering RNAs (siRNAs)-associated stress8,9, by converting superoxide to water. Despite the controversial role of GPX7 in carcinogenesis5,23, there is limited literature on the potential mechanisms of GPX7 expression in gliomas. Herein, our findings provide insights into understanding the epigenetic regulation and the potential association of GPX7 with clinical features and immunity of gliomas.
In our study, we firstly found up-regulation of GPX7 in a pan-cancer analysis, suggesting that GPX7 might function as an extensive tumor-promoter23,110. Further interrogating glioma tumors from multiple cohorts, we found that this peroxidase was markedly higher as pathological grade increased. Its expression was also strongly impacted by other clinicopathological and molecular characteristics, which conferred a worse outcome. These results, along with a recent report111 provide firm evidence of the oncogenic role of GPX7 in gliomas, pointing out its crucial activity in tumor progression and survival.
There is a growing awareness that GPX7 deficiency increases sensitivity to oxidative stress. However, the pathways regulating this process remain elusive, especially in the context of gliomas. Although GPX7 depletion is non-lethal, GPx7−/− mice carry multi-organ abnormalities (e.g. cardiomegaly, splenomegaly, glomerulonephritis and fatty liver) due to increased systemic oxidative stress damage, in addition to increased risk of carcinogenesis, severe oxidative DNA damage and reduced lifespan10. Similarly, it is also known that GPx7-deficient cells accumulate endogenous ROS, lowering cellular viability8,10, and especially in breast cancer cells, GPX7 is essential for reducing the oxidative stress generated by specific polyunsaturated fatty acids112. Furthermore, we now have evidence that mutations in TP53 and EGFR attenuate ROS accumulation in cancer cells by sustaining an oncogenic oxidant intracellular environment through an integrated regulation of redox-related enzymes and signaling pathways (e.g. PI3K-AKT signaling, MAPK cascade, ERK and the redox-sensitive IκK/NF-κB pathway)113,114, thereby supporting proliferation, protein synthesis and invasion106,115,116,117,118. In line with this, we demonstrated that GPX7 was overexpressed in glioma cell lines in a cell cycle-dependent manner and was higher in patients harboring mutations in TP53 and EGFR, suggesting its involvement in cancer cell growth associated with a response to a progressive accumulation of ROS, probably driven by mutant TP53 and EGFR combined with a metabolic reconfiguration and a coordinated regulation of other factors present in the glioma TME (e.g. hypoxia, inflammatory cytokines, growth factors)109,119.
In addition, GPX7 expression increased in chemotherapy-resistant GBM patients, supporting the idea that changes in GPX7 expression are likely to be related to the induction of oxidative stress and ROS-mediated cell injury induced by specific drugs approved to treat gliomas. The results of many studies support a scenario in which increased ROS levels control the multidrug resistance of cancer cells in multiple ways, which leads to development and metastasis during or after chemotherapy. Such resistance is the result of a process known as “Redox Resetting”120,121, where a new redox balance is established with a higher ROS level through the upregulation of the ROS-scavenging system, such as GPX7, which can confer drug resistance118,122,123,124. Although our results suggest the importance of GPX7 in relieving oxidative stress in gliomas, it becomes of paramount importance to reveal the molecular mechanisms underlying the redox status in this type of cancer, since they might potentially open the discovery of appropriate strategies to selectively modulate ROS for the “oxidation therapy”.
Tumorigenesis is due to the combined action of multiple epigenetic events, such as DNA methylation, histone modifications, chromatin remodeling and microRNAs (miRNAs)125. In light of this evidence, it is not surprising that these epigenetics processes have been reported to be involved with every aspect of pathophysiology, diagnosis, and treatment of gliomas126,127,128. However, the epigenetic mechanisms that potentially modulate GPX7 expression in gliomas are still unknown. Our analysis established that DNA hypomethylation in the CpG islands and shores may be one of the mechanisms in leading to GPX7 overexpression in LGG, agreeing with Peng et al.21 and Chen et al.22, who also reported the same negative correlation when analyzing the promoter region of the GPX7 in esophageal adenocarcinomas and gastric cancer, respectively. Considering that ROS impacts the activities of epigenetic modulators129,130,131, it is reasonable to hypothesize that high levels of ROS generated by increased metabolic rate, gene mutation and relative hypoxia117 perhaps explain the DNA hypomethylation and GPX7 overexpression seen in gliomas. This is presumably due in part to ROS-induced oxidation of adenine (8-oxo-A) and guanine (8-oxo-G), which impairs DNA methylation patterns, since damaged bases in the nascent DNA strand can either inhibit the methylation of cytosine within a distance of 1–2 bp (base pairs) or the binding to the methyltransferase, thereby leading to global hypomethylation132,133,134,135.
Moving beyond the DNA methylation regulation, we found a close correlation between GPX7 expression with the deposition of active histone modifications, which may support a crosstalk model between DNA hypomethylation and the H3K9ac and H3K27ac deposition at active regulatory elements and at the TSS region, which establish a chromatin conformation that is compatible with GPX7 expression136. Indeed, it has been shown that in cancer cells, the accumulation and spreading of H3K27ac enhance oncogene expression, and the chromosomal rearrangements and genetic alterations of HAT activity can also influence the frequency of interactions of these chromatin structures, thereby favoring gene expression137. Accordingly, DNA hypomethylation and/or reduced H3K27me3 point to be the major driving forces of activated gene expression in K27M mutant pediatric high-grade gliomas (pHGGs)138. As such, H3.3K27M mutation accompanied by increased total H3K27ac and reduction in H3K27me3, leading to glioma formation and subsequent tumor progression in a brainstem glioma model139. Lastly, another potential insight into the epigenetic mechanisms related to GPX7 expression comes from our co-expression analyses showing that specific miRNA signatures can regulate GPX7 in gliomas, concurrent with a previous report wherein miR-137 and miR-29b were shown to bind to the 3′ UTR region of GPX7 and inhibit its expression in both SW480 (human colon adenocarcinoma) and HEK293 (human embryonic kidney cell line) cell lines140. Thus, the functional relationship between GPX7 expression and epigenetic modifications is complex and further mechanistic studies are required to explain this dependency.
We next explored the genes significantly associated with GPX7 and their functions in gliomas. Conceivably, our data indicated that the functions of GPX7 and associated genes were primarily involved in immune mechanisms in LGG. In contrast, in GBM, they were related to the metabolic regulation of mitochondrial dynamics. Curiously, a recent pathway-based classification suggested that GBM tumors classified as mitochondrial exhibited marked vulnerability to inhibitors of oxidative phosphorylation (OXPHOS), which increased intracellular ROS and sensitivity to radiotherapy, thus strengthening the potential application of synergic GPX7 and OXPHOS inhibition in mitochondrial subtype to boost antitumor and immune responses141.
Importantly, this study consistently showed that GPX7 expression has a dramatic association with immune infiltration and the degree of activation of diverse immune cells using different algorithms. Furthermore, this association was tumor-type dependent, especially prominent in LGG but not in GBM. Interestingly, GPX7 expression and other enzymes associated with redox balance were also increased in other inflammatory contexts, such as chronic HCV infection20, suggesting that this pathway may be upregulated as a feedback mechanism in these contexts to cope with oxidative stress generated in these conditions. Therefore, upregulation of GPX7 observed in gliomas might reflect the oxidative stress that these tumors experience during their development, as supported by its positive correlation with the cell cycle, chemotherapy treatment, and inflammatory pathways and cells. Prominent among these cells, neutrophils, CD4+ T cells, mDCs and CAFs, along with the GPX7 expression, exerted a significant negative influence on the OS of LGG, suggesting once again that both GPX7 and immune cell infiltration increase along with tumor aggressiveness, probably driven by increased oxidative stress in the tumor microenvironment. Of note, GBMs are known to be poorly immunogenic due to active production of immunomodulatory molecules by tumor cells142, which conceivably explains the lack of association between GPX7 and inflammation in these tumors.
Most glioma patients have a strong neutrophilia143,144, and neutrophil infiltration at the tumor site has been associated with decreased overall survival (OS), tumor recurrence in grade 2–4 glioma patients and brain metastasis145. Circulating neutrophils are recruited at the tumor site by CXCL8 produced by FasL triggering on glioma cells146,147 or by the Migration Inhibitory Factor (MIF) produced by glioma cancer stem cells148. At the tumoral site, tumor-associated neutrophils (TANs) support glioma infiltration and progression, respectively, secreting elastase149 and neutrophil extracellular traps (NETs)150, which regulate the HMGB1/RAGE/IL-8 axis150, that ultimately leads to ROS generation, thus partially explaining, therefore, the upregulation of GPX7 associated with neutrophil infiltration seen in our study. Additionally, our results align well with the involvement of CCL2/5, CCR2, and CXCR2/4 in regulating the TAN homing to glioma microenvironment, as we identified striking positive correlations of GPX7 with these chemokines/receptors151,152,153, thus revealing the potential role of GPX7 in the mobilization and recruitment of TANs through this pathway.
The role of CD4+ T lymphocytes is increasingly being studied in the T-cell response against tumors. High infiltration of CD4+ T cells is generally considered to be associated with angiogenesis promotion154, recurrence/progression154, and unfavorable prognosis of gliomas154,155. Similarly, our study highlights that LGG patients with high GPX7 expression and increased frequency of CD4+ T cells have worse OS. Together with the extensive epigenetic reprogramming dictated by the glioma TME, the prominent association between GPX7 expression and CD4+ cell subtype signatures (e.g., Th1, Th2 and Treg) seen in our study suggests the role of GPX7 in CD4+ T cell polarization, which may participate in the lack of effective immune activation against gliomas156,157. The results mentioned above, along with the strong association between GPX7 and several immunosuppressive cells (e.g., NKT cells, TAMs, and MDSCs) or inhibitory immune checkpoint markers (e.g., PD-1, CTLA-4, LAG-3, TIGIT, TIM-3 and CD96) further enhances the importance of GPX7 in the regulation of an immunosuppressive tumor microenvironment158,159,160,161,162.
Several studies have highlighted the suppressive impact of TIGIT on a wide range of immune functions and immune cells (e.g., T/NK cells)163,164,165. Besides, TIGIT+ Treg cells selectively inhibit pro-inflammatory Th1 and Th17 cell responses, but not Th2 cell responses166. Interleukin 4 (IL-4) produced by Th2 cells promotes the differentiation of TAM to M2 macrophages, leading to an immunosuppressive phenotype. Interestingly, there is some circumstantial evidence that M2 macrophages require ROS from TME for proper polarization and acquisition of the pro-tumorigenic phenotype167. ROS induces its polarization via IL4-induced Stat3 activation167, up-regulation of PD-L1, mediated by the NF-κB, secretion of immunosuppressive cytokines168, and the differential expression of ROS scavenging enzymes, such as GPx. Our data report a strong correlation between GPX7 expression with the M2 macrophages markers (CD163, VSIG4, MS4A4A), implying a potential role for GPX7 in the polarization of tumor-associated macrophages (TAM) in LGG169.
mDCs are essential players in coordinating the activation of both CD4+ and CD8+ T cells and initiate adaptive immune responses in infectious contexts by capturing and presenting antigens from inflamed tissues to T cells in the lymph nodes170. However, a failure in mDC activation in microenvironments dominated by immunomodulatory molecules (e.g., TGFβ and IL-10) and cells (such as Tregs) can promote a mDC tolerogenic phenotype, inducing T cell anergy upon antigen presentation, which is a frequent process in cancer, especially in well-known cold tumors with decreased mutational burden171, as is the case for gliomas172. Finally, cancer-associated fibroblasts (CAFs) can be activated by ROS and inflammation in the tumor microenvironment173 and are also consistently implicated in tumor progression by supporting tumor growth and invasion through secretion of cytokines and extracellular matrix remodeling components174. Among the CAF-derived cytokines, TGFβ can be highlighted by its effects in inducing EMT and maintenance of stemness in cancer cells, as well as by its immunosuppressive roles175,176. Interestingly, CAFs were shown to induce a suppressive phenotype in dendritic cells that was dependent on ROS generation in the lung cancer microenvironment, suggesting a mechanism that connects CAFs, mDCs and ROS to cancer immunosuppression and progression177.
In light of these data, it is reasonable to suggest that inflammation and ROS generation are interconnected in a feedforward loop during tumor development, triggering GPX7 expression. Notably, the negative impact of neutrophils, mDCs, CD4+ T cells and CAFs associated with increased GPX7 in LGG prognosis suggests that ROS-associated inflammation and immune cell infiltration in these cancers are associated with tumor progression rather than elimination, possibly due to immunosuppressive mechanisms operating in the TME, which might also be induced by oxidative stress. The biology of GPX7 and its effects in multiple cell types and pathologic contexts are only beginning to be dissected, and further mechanistic studies investigating GPX7 roles in cancer and immunity might reveal more complex mechanisms.
In conclusion, we have shown that GPX7 overexpression has an oncogenic role and was related to worse clinical evolution in gliomas. Importantly, we showed that this increase was associated with greater histological or molecular features of glioma malignancy, and we suggest that this upregulation might be related to a progressive accumulation of ROS in the TME. Notably, we provided the first evidence regarding the epigenetic-mediated regulatory mechanisms underlying GPX7 activation in gliomas. Furthermore, our study yields critical insights into the significant effect of GPX7 in modulating the immune molecules and immune cell infiltration in the microenvironment of gliomas, impacting patient outcomes, opening up future opportunities to regulate the immune response.
Data availability
The datasets analyzed for this study can be found in the Chinese Glioma Genome Atlas (CGGA) (http://www.cgga.org.cn/), GEPIA (http://gepia.cancer-pku.cn/index.html), GlioVis (http://gliovis.bioinfo.cnio.es/), Oncomine (http://www.oncomine.org), TISIDB (http://cis.hku.hk/TISIDB), Tumor Immune Estimation Resource (TIMER, https://cistrome.shinyapps.io/timer), TCGA databases (https://tcga-data.nci.nih.gov/tcga/), Cancer Cell Line Encyclopedia (CCLE, https://portals.broadinstitute.org/ccle/), The Gene Expression Omnibus (GEO) (https://www.ncbi.nlm.nih.gov/gds) and University of California Santa Cruz (UCSC) Xena Browser (https://xena.ucsc.edu/). Further inquiries can be directed to the corresponding author.
Abbreviations
- BEV:
-
Bevacizumab
- CNA:
-
Copy number alteration
- CGGA:
-
Chinese Glioma Genome Atlas
- DFS:
-
Disease-free survival
- GBM:
-
Glioblastoma
- GPX7 :
-
Glutathione peroxidase 7
- HAT:
-
Histone acetyltransferase
- HGGs:
-
High-grade gliomas
- LGG:
-
Low-grade gliomas
- OS:
-
Overall survival
- ROS:
-
Reactive oxygen species
- TCGA:
-
The Cancer Genome Atlas
- TME:
-
Tumor microenvironment
- TMZ:
-
Temozolomide
References
Ostrom, Q. T. et al. CBTRUS Statistical report: Primary brain and other central nervous system tumors diagnosed in the United States in 2013–2017. Neuro-oncology 22, iv1-iv96. https://doi.org/10.1093/neuonc/noaa200 (2020).
Louis, D. N. et al. The 2021 WHO classification of tumors of the central nervous system: A summary. Neuro Oncol. 23, 1231–1251. https://doi.org/10.1093/neuonc/noab106 (2021).
Rushing, E. J. WHO classification of tumors of the nervous system: preview of the upcoming 5th edition. Memo-Mag. Eur. Med. Oncol. 1–4 (2021).
Kan, L. K. et al. Potential biomarkers and challenges in glioma diagnosis, therapy and prognosis. BMJ Neurol. Open 2, e000069. https://doi.org/10.1136/bmjno-2020-000069 (2020).
Kanemura, S. et al. Characterization of the endoplasmic reticulum-resident peroxidases GPx7 and GPx8 shows the higher oxidative activity of GPx7 and its linkage to oxidative protein folding. J. Biol. Chem. 295, 12772–12785. https://doi.org/10.1074/jbc.RA120.013607 (2020).
Wang, L., Zhang, L., Niu, Y., Sitia, R. & Wang, C. C. Glutathione peroxidase 7 utilizes hydrogen peroxide generated by Ero1alpha to promote oxidative protein folding. Antioxid. Redox Signal. 20, 545–556. https://doi.org/10.1089/ars.2013.5236 (2014).
Buday, K. & Conrad, M. Emerging roles for non-selenium containing ER-resident glutathione peroxidases in cell signaling and disease. Biol. Chem. 402, 271–287. https://doi.org/10.1515/hsz-2020-0286 (2021).
Chen, Y. I., Wei, P. C., Hsu, J. L., Su, F. Y. & Lee, W. H. NPGPx (GPx7): A novel oxidative stress sensor/transmitter with multiple roles in redox homeostasis. Am. J. Transl. Res. 8, 1626–1640 (2016).
Wei, P. C., Lo, W. T., Su, M. I., Shew, J. Y. & Lee, W. H. Non-targeting siRNA induces NPGPx expression to cooperate with exoribonuclease XRN2 for releasing the stress. Nucleic Acids Res. 40, 323–332. https://doi.org/10.1093/nar/gkr714 (2012).
Wei, P. C. et al. Loss of the oxidative stress sensor NPGPx compromises GRP78 chaperone activity and induces systemic disease. Mol. Cell 48, 747–759. https://doi.org/10.1016/j.molcel.2012.10.007 (2012).
Muthukumar, K. & Nachiappan, V. Cadmium-induced oxidative stress in Saccharomyces cerevisiae. Indian J. Biochem. Biophys. 47, 383–387 (2010).
Chang, Y. C. et al. Deficiency of NPGPx, an oxidative stress sensor, leads to obesity in mice and human. EMBO Mol. Med. 5, 1165–1179. https://doi.org/10.1002/emmm.201302679 (2013).
Nguyen, V. D. et al. Two endoplasmic reticulum PDI peroxidases increase the efficiency of the use of peroxide during disulfide bond formation. J. Mol. Biol. 406, 503–515. https://doi.org/10.1016/j.jmb.2010.12.039 (2011).
Chen, P. J. et al. NPGPx modulates CPEB2-controlled HIF-1alpha RNA translation in response to oxidative stress. Nucleic Acids Res. 43, 9393–9404. https://doi.org/10.1093/nar/gkv1010 (2015).
Fang, J. et al. Metformin alleviates human cellular aging by upregulating the endoplasmic reticulum glutathione peroxidase 7. Aging Cell 17, e12765. https://doi.org/10.1111/acel.12765 (2018).
Bannister, C. A. et al. Can people with type 2 diabetes live longer than those without? A comparison of mortality in people initiated with metformin or sulphonylurea monotherapy and matched, non-diabetic controls. Diabetes Obes. Metab. 16, 1165–1173. https://doi.org/10.1111/dom.12354 (2014).
Hsieh, Y. L. et al. NPGPx-mediated adaptation to oxidative stress protects motor neurons from degeneration in aging by directly modulating O-GlcNAcase. Cell Rep. 29, 2134–2143 e2137. https://doi.org/10.1016/j.celrep.2019.10.053 (2019).
Groves, J. A., Lee, A., Yildirir, G. & Zachara, N. E. Dynamic O-GlcNAcylation and its roles in the cellular stress response and homeostasis. Cell Stress Chaperones 18, 535–558. https://doi.org/10.1007/s12192-013-0426-y (2013).
Hart, G. W., Slawson, C., Ramirez-Correa, G. & Lagerlof, O. Cross talk between O-GlcNAcylation and phosphorylation: Roles in signaling, transcription, and chronic disease. Annu. Rev. Biochem. 80, 825–858. https://doi.org/10.1146/annurev-biochem-060608-102511 (2011).
Shahid, M. et al. Blood-based gene expression profile of oxidative stress and antioxidant genes for identifying surrogate markers of liver tissue injury in chronic hepatitis C patients. Arch. Virol. 165, 809–822. https://doi.org/10.1007/s00705-020-04564-z (2020).
Peng, D. et al. Glutathione peroxidase 7 has potential tumour suppressor functions that are silenced by location-specific methylation in oesophageal adenocarcinoma. Gut 63, 540–551. https://doi.org/10.1136/gutjnl-2013-304612 (2014).
Chen, Z. et al. Glutathione peroxidase 7 suppresses cancer cell growth and is hypermethylated in gastric cancer. Oncotarget 8, 54345–54356. https://doi.org/10.18632/oncotarget.17527 (2017).
Guerriero, E. et al. GPX4 and GPX7 over-expression in human hepatocellular carcinoma tissues. Eur. J. Histochem. EJH 59, 2540. https://doi.org/10.4081/ejh.2015.2540 (2015).
Wei, J. et al. Identification the prognostic value of glutathione peroxidases expression levels in acute myeloid leukemia. Ann. Transl. Med. 8, 678. https://doi.org/10.21037/atm-20-3296 (2020).
Rusolo, F. et al. Comparison of the seleno-transcriptome expression between human non-cancerous mammary epithelial cells and two human breast cancer cell lines. Oncol. Lett. 13, 2411–2417. https://doi.org/10.3892/ol.2017.5715 (2017).
Rhodes, D. R. et al. ONCOMINE: A cancer microarray database and integrated data-mining platform. Neoplasia 6, 1–6. https://doi.org/10.1016/s1476-5586(04)80047-2 (2004).
Madhavan, S. et al. Rembrandt: Helping personalized medicine become a reality through integrative translational research. Mol. Cancer Res. MCR 7, 157–167. https://doi.org/10.1158/1541-7786.MCR-08-0435 (2009).
Gravendeel, L. A. et al. Intrinsic gene expression profiles of gliomas are a better predictor of survival than histology. Can. Res. 69, 9065–9072. https://doi.org/10.1158/0008-5472.CAN-09-2307 (2009).
Kamoun, A. et al. Integrated multi-omics analysis of oligodendroglial tumours identifies three subgroups of 1p/19q co-deleted gliomas. Nat. Commun. 7, 11263. https://doi.org/10.1038/ncomms11263 (2016).
Zhao, Z. et al. Chinese Glioma Genome Atlas (CGGA): A comprehensive resource with functional genomic data from Chinese glioma patients. Genomics Proteomics Bioinform. 19, 1–12. https://doi.org/10.1016/j.gpb.2020.10.005 (2021).
Bowman, R. L., Wang, Q., Carro, A., Verhaak, R. G. & Squatrito, M. GlioVis data portal for visualization and analysis of brain tumor expression datasets. Neuro Oncol. 19, 139–141. https://doi.org/10.1093/neuonc/now247 (2017).
Ghandi, M. et al. Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature 569, 503–508. https://doi.org/10.1038/s41586-019-1186-3 (2019).
Nusinow, D. P. et al. Quantitative proteomics of the Cancer Cell Line Encyclopedia. Cell 180, 387–402 e316. https://doi.org/10.1016/j.cell.2019.12.023 (2020).
Clough, E. & Barrett, T. The gene expression omnibus database. Methods Mol. Biol. 1418, 93–110. https://doi.org/10.1007/978-1-4939-3578-9_5 (2016).
Litovchick, L. et al. Evolutionarily conserved multisubunit RBL2/p130 and E2F4 protein complex represses human cell cycle-dependent genes in quiescence. Mol. Cell 26, 539–551 (2007).
Huang, G. et al. The microarray gene profiling analysis of glioblastoma cancer cells reveals genes affected by FAK inhibitor Y15 and combination of Y15 and temozolomide. Anticancer Agents Med. Chem. 14, 9–17. https://doi.org/10.2174/18715206113139990141 (2014).
Masiero, M. et al. A core human primary tumor angiogenesis signature identifies the endothelial orphan receptor ELTD1 as a key regulator of angiogenesis. Cancer Cell 24, 229–241. https://doi.org/10.1016/j.ccr.2013.06.004 (2013).
Mueller, W. et al. Downregulation of RUNX3 and TES by hypermethylation in glioblastoma. Oncogene 26, 583–593. https://doi.org/10.1038/sj.onc.1209805 (2007).
Tang, Z., Kang, B., Li, C., Chen, T. & Zhang, Z. GEPIA2: An enhanced web server for large-scale expression profiling and interactive analysis. Nucleic Acids Res. 47, W556–W560. https://doi.org/10.1093/nar/gkz430 (2019).
Tang, Z. et al. GEPIA: A web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 45, W98–W102. https://doi.org/10.1093/nar/gkx247 (2017).
Zhao, Z. et al. Chinese Glioma Genome Atlas (CGGA): A comprehensive resource with functional genomic data from Chinese gliomas. Genomics Proteomics Bioinform. 19, 1–12 (2021).
Clark, K. et al. The Cancer Imaging Archive (TCIA): Maintaining and operating a public information repository. J. Digit. Imaging 26, 1045–1057 (2013).
Scarpace, L., Flanders, A., Jain, R., Mikkelsen, T. & Andrews, D. W. (Published, 2015).
Scarpace, L., Flanders, A. E., Jain, R., Mikkelsen, T. & Andrews, D. W. Data from REMBRANDT. The Cancer Imaging Archive 10, K9 (2015).
Fekete, J. T. & Gyorffy, B. ROCplot.org: Validating predictive biomarkers of chemotherapy/hormonal therapy/anti-HER2 therapy using transcriptomic data of 3,104 breast cancer patients. Int. J. Cancer 145, 3140–3151. https://doi.org/10.1002/ijc.32369 (2019).
Vasaikar, S., Straub, P., Wang, J. & Zhang, B. (AACR, 2019).
Vasaikar, S. V., Straub, P., Wang, J. & Zhang, B. LinkedOmics: analyzing multi-omics data within and across 32 cancer types. Nucleic Acids Res. 46, D956–D963 (2018).
Modhukur, V. et al. MethSurv: A web tool to perform multivariable survival analysis using DNA methylation data. Epigenomics 10, 277–288. https://doi.org/10.2217/epi-2017-0118 (2018).
Metsalu, T. & Vilo, J. ClustVis: A web tool for visualizing clustering of multivariate data using Principal Component Analysis and heatmap. Nucleic Acids Res. 43, W566-570. https://doi.org/10.1093/nar/gkv468 (2015).
Li, Y., Ge, D. & Lu, C. The SMART App: An interactive web application for comprehensive DNA methylation analysis and visualization. Epigenetics Chromatin 12, 71. https://doi.org/10.1186/s13072-019-0316-3 (2019).
Vlachos, I. S. et al. DIANA-miRPath v3.0: Deciphering microRNA function with experimental support. Nucleic Acids Res. 43, W460-466. https://doi.org/10.1093/nar/gkv403 (2015).
Friedman, R. C., Farh, K. K., Burge, C. B. & Bartel, D. P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 19, 92–105. https://doi.org/10.1101/gr.082701.108 (2009).
Dweep, H., Sticht, C., Pandey, P. & Gretz, N. miRWalk–database: Prediction of possible miRNA binding sites by “walking” the genes of three genomes. J. Biomed. Inform. 44, 839–847 (2011).
Dweep, H., Gretz, N. & Sticht, C. in RNA Mapping 289–305 (Springer, 2014).
Chen, Y. & Wang, X. miRDB: an online database for prediction of functional microRNA targets. Nucleic Acids Res. 48, D127–D131 (2020).
Wang, X. miRDB: A microRNA target prediction and functional annotation database with a wiki interface. RNA 14, 1012–1017 (2008).
Wong, N. & Wang, X. miRDB: An online resource for microRNA target prediction and functional annotations. Nucleic Acids Res. 43, D146–D152 (2015).
Du, J. et al. KEGG-PATH: Kyoto encyclopedia of genes and genomes-based pathway analysis using a path analysis model. Mol. BioSyst. 10, 2441–2447 (2014).
Kanehisa, M. KEGG: Kyoto encyclopedia of genes and genomes. Kanehisa laboratories (2012).
Kanehisa, M., Furumichi, M., Sato, Y., Ishiguro-Watanabe, M. & Tanabe, M. KEGG: Integrating viruses and cellular organisms. Nucleic Acids Res. 49, D545–D551. https://doi.org/10.1093/nar/gkaa970 (2021).
Li, T. et al. TIMER2.0 for analysis of tumor-infiltrating immune cells. Nucleic Acids Res. 48, W509–W514. https://doi.org/10.1093/nar/gkaa407 (2020).
Li, T. et al. TIMER: A web server for comprehensive analysis of tumor-infiltrating immune cells. Can. Res. 77, e108–e110. https://doi.org/10.1158/0008-5472.CAN-17-0307 (2017).
Ru, B. et al. TISIDB: An integrated repository portal for tumor-immune system interactions. Bioinformatics 35, 4200–4202. https://doi.org/10.1093/bioinformatics/btz210 (2019).
Gutmann, D. H. et al. Comparative gene expression profile analysis of neurofibromatosis 1-associated and sporadic pilocytic astrocytomas. Can. Res. 62, 2085–2091 (2002).
Sun, L. et al. Neuronal and glioma-derived stem cell factor induces angiogenesis within the brain. Cancer Cell 9, 287–300. https://doi.org/10.1016/j.ccr.2006.03.003 (2006).
Murat, A. et al. Stem cell-related “self-renewal” signature and high epidermal growth factor receptor expression associated with resistance to concomitant chemoradiotherapy in glioblastoma. J. Clin. Oncol. 26, 3015–3024. https://doi.org/10.1200/JCO.2007.15.7164 (2008).
Shai, R. et al. Gene expression profiling identifies molecular subtypes of gliomas. Oncogene 22, 4918–4923. https://doi.org/10.1038/sj.onc.1206753 (2003).
French, P. J. et al. Gene expression profiles associated with treatment response in oligodendrogliomas. Can. Res. 65, 11335–11344. https://doi.org/10.1158/0008-5472.CAN-05-1886 (2005).
Patel, A. P. et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 344, 1396–1401. https://doi.org/10.1126/science.1254257 (2014).
Markouli, M., Strepkos, D., Papavassiliou, K. A., Papavassiliou, A. G. & Piperi, C. Bivalent genes targeting of glioma heterogeneity and plasticity. Int. J. Mol. Sci. https://doi.org/10.3390/ijms22020540 (2021).
Shiras, A. & Mondal, A. Extrachromosomal DNA: Redefining the pathogenesis of glioblastoma. Biochim. Biophys. Acta Rev. Cancer 1876, 188551. https://doi.org/10.1016/j.bbcan.2021.188551 (2021).
Lyon, J. F. et al. Spatial progression and molecular heterogeneity of IDH-mutant glioblastoma determined by DNA methylation-based mapping. Acta Neuropathol. Commun. 9, 120. https://doi.org/10.1186/s40478-021-01221-7 (2021).
Law, M. E. et al. Molecular cytogenetic analysis of chromosomes 1 and 19 in glioma cell lines. Cancer Genet. Cytogenet. 160, 1–14. https://doi.org/10.1016/j.cancergencyto.2004.11.012 (2005).
Bigner, S. H. & Schrock, E. Molecular cytogenetics of brain tumors. J. Neuropathol. Exp. Neurol. 56, 1173–1181. https://doi.org/10.1097/00005072-199711000-00001 (1997).
Goussia, A. C., Agnantis, N. J., Rao, J. S. & Kyritsis, A. P. Cytogenetic and molecular abnormalities in astrocytic gliomas (Review). Oncol. Rep. 7, 401–412. https://doi.org/10.3892/or.7.2.401 (2000).
Krupp, W., Geiger, K., Schober, R., Siegert, G. & Froster, U. G. Cytogenetic and molecular cytogenetic analyses in diffuse astrocytomas. Cancer Genet. Cytogenet. 153, 32–38. https://doi.org/10.1016/j.cancergencyto.2003.12.017 (2004).
Mao, X. & Hamoudi, R. A. Molecular and cytogenetic analysis of glioblastoma multiforme. Cancer Genet. Cytogenet. 122, 87–92. https://doi.org/10.1016/s0165-4608(00)00278-8 (2000).
Squire, J. A. et al. Molecular cytogenetic analysis of glial tumors using spectral karyotyping and comparative genomic hybridization. Mol. Diagnosis 6, 93–108. https://doi.org/10.1054/modi.2001.22745 (2001).
Fults, D. et al. Molecular cytogenetic analysis of a t(7;10) in a human glioblastoma cell line. Cancer Genet. Cytogenet. 81, 118–124. https://doi.org/10.1016/0165-4608(94)00265-d (1995).
Feuerstein, B. G. & Mohapatra, G. Molecular cytogenetic quantitation of gains and losses of genetic material from human gliomas. J. Neurooncol. 24, 47–55. https://doi.org/10.1007/BF01052658 (1995).
Louis, D. N. et al. The 2016 World Health Organization classification of tumors of the central nervous system: a summary. Acta Neuropathol. 131, 803–820 (2016).
Verhaak, R. G. et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 17, 98–110 (2010).
Abedalthagafi, M., Mobark, N., Al-Rashed, M. & AlHarbi, M. Epigenomics and immunotherapeutic advances in pediatric brain tumors. NPJ Precis. Oncol. 5, 34. https://doi.org/10.1038/s41698-021-00173-4 (2021).
Campos, E. I. & Reinberg, D. Histones: Annotating chromatin. Annu. Rev. Genet. 43, 559–599. https://doi.org/10.1146/annurev.genet.032608.103928 (2009).
Marmorstein, R. & Roth, S. Y. Histone acetyltransferases: Function, structure, and catalysis. Curr. Opin. Genet. Dev. 11, 155–161 (2001).
Neganova, M. E., Klochkov, S. G., Aleksandrova, Y. R. & Aliev, G. in Seminars in cancer biology. (Elsevier).
Narita, T., Weinert, B. T. & Choudhary, C. Functions and mechanisms of non-histone protein acetylation. Nat. Rev. Mol. Cell Biol. 20, 156–174 (2019).
Neganova, M. E., Klochkov, S. G., Aleksandrova, Y. R. & Aliev, G. Histone modifications in epigenetic regulation of cancer: Perspectives and achieved progress. Semin. Cancer Biol. https://doi.org/10.1016/j.semcancer.2020.07.015 (2020).
Chen, X. et al. A novel enhancer regulates MGMT expression and promotes temozolomide resistance in glioblastoma. Nat. Commun. 9, 1–14 (2018).
Wang, X. et al. Purine synthesis promotes maintenance of brain tumor initiating cells in glioma. Nat. Neurosci. 20, 661–673 (2017).
Kalinin, V. Cell–extracellular matrix interaction in glioma growth. In silico model. J. Integr. Bioinform. https://doi.org/10.1515/jib-2020-0027 (2020).
Sampson, J. H., Gunn, M. D., Fecci, P. E. & Ashley, D. M. Brain immunology and immunotherapy in brain tumours. Nat. Rev. Cancer 20, 12–25 (2020).
Touat, M. et al. Mechanisms and therapeutic implications of hypermutation in gliomas. Nature 580, 517–523 (2020).
Stupp, R. & Mason, W. Bent MJ van den et al. on behalf of the European Organisation for Research and Treatment of Cancer (EORTC) Brain Tumor and Radiotherapy Groups and National Cancer Institute of Canada Clinical Trials Group (NCIC CTG) (2005) Radiotherapy plus concomitant and adjuvant temozolomide for patients with newly diagnosed glioblastoma. New Engl. J. Med. 352, 987–996.
van den Bent, M. J. et al. Adjuvant procarbazine, lomustine, and vincristine chemotherapy in newly diagnosed anaplastic oligodendroglioma: Long-term follow-up of EORTC brain tumor group study 26951. J. Clin. Oncol. 31, 344–350 (2013).
Seystahl, K. et al. Bevacizumab versus alkylating chemotherapy in recurrent glioblastoma. J. Cancer Res. Clin. Oncol. 146, 659–670 (2020).
Lu, G. et al. Triple-drug therapy with bevacizumab, irinotecan, and temozolomide plus tumor treating fields for recurrent glioblastoma: A retrospective study. Front. Neurol. 10, 42 (2019).
Jakobsen, J. et al. Toxicity and efficacy of lomustine and bevacizumab in recurrent glioblastoma patients. J. Neurooncol. 137, 439–446 (2018).
Burger, M. C. et al. Bevacizumab for patients with recurrent multifocal glioblastomas. Int. J. Mol. Sci. 18, 2469 (2017).
Sever, O. N. et al. Reoperation does not provide a survival advantage in patients with recurrent Glioblastoma treated with irinotecan/bevacizumab treatment. Indian J. Cancer 58, 91 (2021).
Yang, H. et al. The role of cellular reactive oxygen species in cancer chemotherapy. J. Exp. Clin. Cancer Res. 37, 1–10 (2018).
Feng, Q., Li, L., Li, M. & Wang, X. Immunological classification of gliomas based on immunogenomic profiling. J. Neuroinflammation 17, 360. https://doi.org/10.1186/s12974-020-02030-w (2020).
Zhang, B., Shen, R., Cheng, S. & Feng, L. Immune microenvironments differ in immune characteristics and outcome of glioblastoma multiforme. Cancer Med. 8, 2897–2907. https://doi.org/10.1002/cam4.2192 (2019).
Kohli, K., Pillarisetty, V. G. & Kim, T. S. Key chemokines direct migration of immune cells in solid tumors. Cancer Gene Ther. https://doi.org/10.1038/s41417-021-00303-x (2021).
Waris, G. & Ahsan, H. Reactive oxygen species: Role in the development of cancer and various chronic conditions. J. Carcinogenesis 5, 14. https://doi.org/10.1186/1477-3163-5-14 (2006).
Salazar-Ramiro, A. et al. Role of redox status in development of glioblastoma. Front. Immunol. 7, 156. https://doi.org/10.3389/fimmu.2016.00156 (2016).
Valko, M. et al. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 39, 44–84. https://doi.org/10.1016/j.biocel.2006.07.001 (2007).
Policastro, L. L., Ibanez, I. L., Notcovich, C., Duran, H. A. & Podhajcer, O. L. The tumor microenvironment: Characterization, redox considerations, and novel approaches for reactive oxygen species-targeted gene therapy. Antioxid. Redox Signal. 19, 854–895. https://doi.org/10.1089/ars.2011.4367 (2013).
Reuter, S., Gupta, S. C., Chaturvedi, M. M. & Aggarwal, B. B. Oxidative stress, inflammation, and cancer: how are they linked?. Free Radical Biol. Med. 49, 1603–1616. https://doi.org/10.1016/j.freeradbiomed.2010.09.006 (2010).
Peng, D. F., Hu, T. L., Soutto, M., Belkhiri, A. & El-Rifai, W. Loss of glutathione peroxidase 7 promotes TNF-alpha-induced NF-kappaB activation in Barrett’s carcinogenesis. Carcinogenesis 35, 1620–1628. https://doi.org/10.1093/carcin/bgu083 (2014).
Yao, J. et al. The increasing expression of GPX7 related to the malignant clinical features leading to poor prognosis of glioma patients. Chin. Neurosurg. J. 7, 21. https://doi.org/10.1186/s41016-021-00235-3 (2021).
Utomo, A. et al. Identification of a novel putative non-selenocysteine containing phospholipid hydroperoxide glutathione peroxidase (NPGPx) essential for alleviating oxidative stress generated from polyunsaturated fatty acids in breast cancer cells. J. Biol. Chem. 279, 43522–43529. https://doi.org/10.1074/jbc.M407141200 (2004).
Cordani, M. et al. Mutant p53 blocks SESN1/AMPK/PGC-1alpha/UCP2 axis increasing mitochondrial O2−. production in cancer cells. Br. J. Cancer 119, 994–1008. https://doi.org/10.1038/s41416-018-0288-2 (2018).
Cordani, M. et al. Mutant p53-associated molecular mechanisms of ROS regulation in cancer cells. Biomolecules. https://doi.org/10.3390/biom10030361 (2020).
Bae, Y. S. et al. Epidermal growth factor (EGF)-induced generation of hydrogen peroxide. Role in EGF receptor-mediated tyrosine phosphorylation. J. Biol. Chem. 272, 217–221 (1997).
Miller, E. W., Tulyathan, O., Isacoff, E. Y. & Chang, C. J. Molecular imaging of hydrogen peroxide produced for cell signaling. Nat. Chem. Biol. 3, 263–267. https://doi.org/10.1038/nchembio871 (2007).
Strickland, M. & Stoll, E. A. Metabolic reprogramming in glioma. Front. Cell Dev. Biol. 5, 43. https://doi.org/10.3389/fcell.2017.00043 (2017).
Hayes, J. D., Dinkova-Kostova, A. T. & Tew, K. D. Oxidative stress in cancer. Cancer Cell 38, 167–197. https://doi.org/10.1016/j.ccell.2020.06.001 (2020).
Rinaldi, M. et al. ROS and brain gliomas: An overview of potential and innovative therapeutic strategies. Int. J. Mol. Sci. https://doi.org/10.3390/ijms17060984 (2016).
Liu, Y. et al. Cancer drug resistance: Redox resetting renders a way. Oncotarget 7, 42740–42761. https://doi.org/10.18632/oncotarget.8600 (2016).
Kramer, R. A., Zakher, J. & Kim, G. Role of the glutathione redox cycle in acquired and de novo multidrug resistance. Science 241, 694–697. https://doi.org/10.1126/science.3399900 (1988).
Trachootham, D., Alexandre, J. & Huang, P. Targeting cancer cells by ROS-mediated mechanisms: A radical therapeutic approach?. Nat. Rev. Drug Discov. 8, 579–591. https://doi.org/10.1038/nrd2803 (2009).
Sabharwal, S. S. & Schumacker, P. T. Mitochondrial ROS in cancer: Initiators, amplifiers or an Achilles’ heel?. Nat. Rev. Cancer 14, 709–721. https://doi.org/10.1038/nrc3803 (2014).
Galadari, S., Rahman, A., Pallichankandy, S. & Thayyullathil, F. Reactive oxygen species and cancer paradox: To promote or to suppress?. Free Radic. Biol. Med. 104, 144–164. https://doi.org/10.1016/j.freeradbiomed.2017.01.004 (2017).
Cheng, Y. et al. Targeting epigenetic regulators for cancer therapy: mechanisms and advances in clinical trials. Signal Transduct. Target. Ther. 4, 62. https://doi.org/10.1038/s41392-019-0095-0 (2019).
Kondo, Y., Katsushima, K., Ohka, F., Natsume, A. & Shinjo, K. Epigenetic dysregulation in glioma. Cancer Sci. 105, 363–369. https://doi.org/10.1111/cas.12379 (2014).
Zang, L., Kondengaden, S. M., Che, F., Wang, L. & Heng, X. Potential epigenetic-based therapeutic targets for glioma. Front. Mol. Neurosci. 11, 408. https://doi.org/10.3389/fnmol.2018.00408 (2018).
Ferreira, W. A. et al. An update on the epigenetics of glioblastomas. Epigenomics 8, 1289–1305. https://doi.org/10.2217/epi-2016-0040 (2016).
Matsushima, S. et al. Increased oxidative stress in the nucleus caused by Nox4 mediates oxidation of HDAC4 and cardiac hypertrophy. Circ. Res. 112, 651–663. https://doi.org/10.1161/CIRCRESAHA.112.279760 (2013).
Ushijima, T. Detection and interpretation of altered methylation patterns in cancer cells. Nat. Rev. Cancer 5, 223–231. https://doi.org/10.1038/nrc1571 (2005).
Perillo, B. et al. ROS in cancer therapy: The bright side of the moon. Exp. Mol. Med. 52, 192–203. https://doi.org/10.1038/s12276-020-0384-2 (2020).
Turk, P. W., Laayoun, A., Smith, S. S. & Weitzman, S. A. DNA adduct 8-hydroxyl-2′-deoxyguanosine (8-hydroxyguanine) affects function of human DNA methyltransferase. Carcinogenesis 16, 1253–1255. https://doi.org/10.1093/carcin/16.5.1253 (1995).
Franco, R., Schoneveld, O., Georgakilas, A. G. & Panayiotidis, M. I. Oxidative stress, DNA methylation and carcinogenesis. Cancer Lett. 266, 6–11. https://doi.org/10.1016/j.canlet.2008.02.026 (2008).
Wachsman, J. T. DNA methylation and the association between genetic and epigenetic changes: relation to carcinogenesis. Mutat. Res. 375, 1–8. https://doi.org/10.1016/s0027-5107(97)00003-1 (1997).
Weitzman, S. A., Turk, P. W., Milkowski, D. H. & Kozlowski, K. Free radical adducts induce alterations in DNA cytosine methylation. Proc. Natl. Acad. Sci. USA 91, 1261–1264. https://doi.org/10.1073/pnas.91.4.1261 (1994).
Kundaje, A. et al. Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330 (2015).
Sungalee, S. et al. Histone acetylation dynamics modulates chromatin conformation and allele-specific interactions at oncogenic loci. Nat. Genet. 53, 650–662. https://doi.org/10.1038/s41588-021-00842-x (2021).
Bender, S. et al. Reduced H3K27me3 and DNA hypomethylation are major drivers of gene expression in K27M mutant pediatric high-grade gliomas. Cancer Cell 24, 660–672. https://doi.org/10.1016/j.ccr.2013.10.006 (2013).
Lewis, P. W. et al. Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science 340, 857–861. https://doi.org/10.1126/science.1232245 (2013).
Hanouskova, B. et al. MicroRNAs mediated regulation of glutathione peroxidase 7 expression and its changes during adipogenesis. Biochim. Biophys. Acta Gene Regul. Mech. 1864, 194734. https://doi.org/10.1016/j.bbagrm.2021.194734 (2021).
Garofano, L. et al. Pathway-based classification of glioblastoma uncovers a mitochondrial subtype with therapeutic vulnerabilities. Nat. Cancer 2, 141–156. https://doi.org/10.1038/s43018-020-00159-4 (2021).
Weller, M. et al. Glioma. Nature reviews. Disease Primers 1, 15017. https://doi.org/10.1038/nrdp.2015.17 (2015).
Gabrusiewicz, K. et al. Glioblastoma-infiltrated innate immune cells resemble M0 macrophage phenotype. JCI Insight. https://doi.org/10.1172/jci.insight.85841 (2016).
Fossati, G. et al. Neutrophil infiltration into human gliomas. Acta Neuropathol. 98, 349–354. https://doi.org/10.1007/s004010051093 (1999).
Ma, L., Li, G. & Wei, M. Neutrophil-to-lymphocyte ratio and its changes are related to grade II–IV glioma recurrence. Cancer Manag. Res. 12, 9429–9434. https://doi.org/10.2147/CMAR.S267523 (2020).
Chio, C. C., Wang, Y. S., Chen, Y. L., Lin, S. J. & Yang, B. C. Down-regulation of Fas-L in glioma cells by ribozyme reduces cell apoptosis, tumour-infiltrating cells, and liver damage but accelerates tumour formation in nude mice. Br. J. Cancer 85, 1185–1192. https://doi.org/10.1054/bjoc.2001.2055 (2001).
Hor, W. S., Huang, W. L., Lin, Y. S. & Yang, B. C. Cross-talk between tumor cells and neutrophils through the Fas (APO-1, CD95)/FasL system: Human glioma cells enhance cell viability and stimulate cytokine production in neutrophils. J. Leukoc. Biol. 73, 363–368. https://doi.org/10.1189/jlb.0702375 (2003).
Otvos, B. et al. Cancer stem cell-secreted macrophage migration inhibitory factor stimulates myeloid derived suppressor cell function and facilitates glioblastoma immune evasion. Stem Cells 34, 2026–2039. https://doi.org/10.1002/stem.2393 (2016).
Iwatsuki, K. et al. Elastase expression by infiltrating neutrophils in gliomas. Neurol. Res. 22, 465–468. https://doi.org/10.1080/01616412.2000.11740701 (2000).
Zha, C. et al. Neutrophil extracellular traps mediate the crosstalk between glioma progression and the tumor microenvironment via the HMGB1/RAGE/IL-8 axis. Cancer Biol. Med. 17, 154–168. https://doi.org/10.20892/j.issn.2095-3941.2019.0353 (2020).
Jablonska, J., Wu, C. F., Andzinski, L., Leschner, S. & Weiss, S. CXCR2-mediated tumor-associated neutrophil recruitment is regulated by IFN-beta. Int. J. Cancer 134, 1346–1358. https://doi.org/10.1002/ijc.28551 (2014).
Pahler, J. C. et al. Plasticity in tumor-promoting inflammation: Impairment of macrophage recruitment evokes a compensatory neutrophil response. Neoplasia 10, 329–340. https://doi.org/10.1593/neo.07871 (2008).
Gao, M. et al. TERT mutation is accompanied by neutrophil infiltration and contributes to poor survival in isocitrate dehydrogenase wild-type glioma. Front. Cell Dev. Biol. 9, 654407. https://doi.org/10.3389/fcell.2021.654407 (2021).
Mu, L. et al. CD4+ and perivascular Foxp3+ T cells in glioma correlate with angiogenesis and tumor progression. Front. Immunol. 8, 1451. https://doi.org/10.3389/fimmu.2017.01451 (2017).
Han, S. et al. Tumour-infiltrating CD4(+) and CD8(+) lymphocytes as predictors of clinical outcome in glioma. Br. J. Cancer 110, 2560–2568. https://doi.org/10.1038/bjc.2014.162 (2014).
Bam, M. et al. Genome wide DNA methylation landscape reveals glioblastoma’s influence on epigenetic changes in tumor infiltrating CD4+ T cells. Oncotarget 12, 967–981. https://doi.org/10.18632/oncotarget.27955 (2021).
Gieryng, A., Pszczolkowska, D., Walentynowicz, K. A., Rajan, W. D. & Kaminska, B. Immune microenvironment of gliomas. Lab. Investig. 97, 498–518. https://doi.org/10.1038/labinvest.2017.19 (2017).
Ito, H., Nakashima, H. & Chiocca, E. A. Molecular responses to immune checkpoint blockade in glioblastoma. Nat. Med. 25, 359–361. https://doi.org/10.1038/s41591-019-0385-7 (2019).
Mirzaei, R., Sarkar, S. & Yong, V. W. T cell exhaustion in glioblastoma: Intricacies of immune checkpoints. Trends Immunol. 38, 104–115. https://doi.org/10.1016/j.it.2016.11.005 (2017).
Liu, F. et al. CD96, a new immune checkpoint, correlates with immune profile and clinical outcome of glioma. Sci. Rep. 10, 10768. https://doi.org/10.1038/s41598-020-66806-z (2020).
Weenink, B. et al. Low-grade glioma harbors few CD8 T cells, which is accompanied by decreased expression of chemo-attractants, not immunogenic antigens. Sci. Rep. 9, 14643. https://doi.org/10.1038/s41598-019-51063-6 (2019).
Mi, Y. et al. The emerging role of myeloid-derived suppressor cells in the glioma immune suppressive microenvironment. Front. Immunol. 11, 737. https://doi.org/10.3389/fimmu.2020.00737 (2020).
Stanietsky, N. et al. The interaction of TIGIT with PVR and PVRL2 inhibits human NK cell cytotoxicity. Proc. Natl. Acad. Sci. USA 106, 17858–17863. https://doi.org/10.1073/pnas.0903474106 (2009).
Wang, F. et al. TIGIT expression levels on human NK cells correlate with functional heterogeneity among healthy individuals. Eur. J. Immunol. 45, 2886–2897. https://doi.org/10.1002/eji.201545480 (2015).
Wen, J., Mao, X., Cheng, Q., Liu, Z. & Liu, F. A pan-cancer analysis revealing the role of TIGIT in tumor microenvironment. Sci. Rep. 11, 22502. https://doi.org/10.1038/s41598-021-01933-9 (2021).
Joller, N. et al. Treg cells expressing the coinhibitory molecule TIGIT selectively inhibit proinflammatory Th1 and Th17 cell responses. Immunity 40, 569–581. https://doi.org/10.1016/j.immuni.2014.02.012 (2014).
Griess, B., Mir, S., Datta, K. & Teoh-Fitzgerald, M. Scavenging reactive oxygen species selectively inhibits M2 macrophage polarization and their pro-tumorigenic function in part, via Stat3 suppression. Free Radic. Biol. Med. 147, 48–60. https://doi.org/10.1016/j.freeradbiomed.2019.12.018 (2020).
Roux, C. et al. Reactive oxygen species modulate macrophage immunosuppressive phenotype through the up-regulation of PD-L1. Proc. Natl. Acad. Sci. USA 116, 4326–4335. https://doi.org/10.1073/pnas.1819473116 (2019).
Xu, J., Liu, F., Li, Y. & Shen, L. A 1p/19q Codeletion-associated immune signature for predicting lower grade glioma prognosis. Cell Mol. Neurobiol. https://doi.org/10.1007/s10571-020-00959-3 (2020).
Wang, Y. et al. Dendritic cell biology and its role in tumor immunotherapy. J. Hematol. Oncol. 13(1), 18 (2020).
Garner, H. & de Visser, K. E. Immune crosstalk in cancer progression and metastatic spread: A complex conversation. Nat. Rev. Immunol. 20, 483–497. https://doi.org/10.1038/s41577-019-0271-z (2020).
Weller, M. et al. Glioma 1, 1–18 (2015).
Arcucci, A., Ruocco, M. R., Granato, G., Sacco, A. M. & Montagnani, S. Cancer: An oxidative crosstalk between solid tumor cells and cancer associated fibroblasts. Biomed. Res. Int. 2016, 4502846. https://doi.org/10.1155/2016/4502846 (2016).
Ping, Q. et al. Cancer-associated fibroblasts: Overview, progress, challenges, and directions. Cancer Gene Ther. 28(9), 984–999 (2021).
Calon, A., Tauriello, D. & Batlle, E. in Seminars in cancer biology. 15–22 (Elsevier).
Batlle, E. & Massagué, J. Transforming growth factor-β signaling in immunity and cancer. Immunity 50(4), 924–940 (2019).
Xiang, H. et al. Cancer-associated fibroblasts promote immunosuppression by inducing ROS-generating monocytic MDSCs in lung squamous cell carcinoma. Cancer Immunol. Res. 8(4), 436–450 (2020).
Acknowledgements
The authors are grateful to Instituto Evandro Chagas (Brazil) and Pró-Reitoria de Pesquisa e Pós-Graduação (PROPESP/UFPA, Brazil) for their financial and technical support.
Author information
Authors and Affiliations
Contributions
W.A.S.F. and E.H.C.O.: conceived and designed the study; W.A.S.F.: performed data mining; W.A.S.F.: writing—original draft preparation; E.H.C.O., G.A.F.V. and T.S.M.: review and editing; E.H.C.O.: funding acquisition. All authors contributed to the article and approved the submitted version.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ferreira, W.A.S., Vitiello, G.A.F., da Silva Medina, T. et al. Comprehensive analysis of epigenetics regulation, prognostic and the correlation with immune infiltrates of GPX7 in adult gliomas. Sci Rep 12, 6442 (2022). https://doi.org/10.1038/s41598-022-10114-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-10114-1
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
