
Abstract
Many human diseases ranging from cancer to hereditary disorders are caused by single-nucleotide mutations in critical genes. Repairing these mutations would significantly improve the quality of life for patients with hereditary diseases. However, current procedures for repairing deleterious single-nucleotide mutations are not straightforward, requiring multiple steps and taking several months to complete. In the current study, we aimed to repair pathogenic allele-specific single-nucleotide mutations using a single round of genome editing. Using high-fidelity, site-specific nuclease AsCas12a/Cpf1, we attempted to repair pathogenic single-nucleotide variants (SNVs) in disease-specific induced pluripotent stem cells. As a result, we achieved repair of the Met918Thr SNV in human oncogene RET with the inclusion of a single-nucleotide marker, followed by absolute markerless, scarless repair of the RET SNV with no detected off-target effects. The markerless method was then confirmed in human type VII collagen-encoding gene COL7A1. Thus, using this One-SHOT method, we successfully reduced the number of genetic manipulations required for genome repair from two consecutive events to one, resulting in allele-specific repair that can be completed within 3 weeks, with or without a single-nucleotide marker. Our findings suggest that One-SHOT can be used to repair other types of mutations, with potential beyond human medicine.
Similar content being viewed by others


Introduction
The human genome contains extensive variation, including an estimated 5 × 106 single-nucleotide variants (SNVs) that determine how we look and function, as well as our specific disease tendencies1,2,3. Some SNVs are pathogenic and either directly or indirectly cause hereditary disorders4,5,6 such as multiple endocrine neoplasia type 2B (MEN2B)7 and dystrophic epidermolysis bullosa (DEB)8. MEN2B is an autosomal dominant syndrome characterised by thyroid, adrenal gland and neuronal tumours and skeletal abnormalities. The majority of MEN2B cases (95%) result from a single amino-acid substitution (Met918Thr) in the RET proto-oncogene, which is caused by a pathogenic SNV (RET: c.2753T>C) at the second base of the codon7. DEB is an inherited disease characterised by severe, recurrent skin ulcers and blistering. It is caused by genetic mutations in the human type VII collagen-encoding gene COL7A1, the product of which is an anchoring fibril connecting the epidermis to the dermis8. To model diseases such as these in vitro, disease-specific induced pluripotent stem cells (iPSCs) carrying pathogenic SNVs or other genetic mutations can be obtained from patients9,10,11,12. Repairing these iPSCs to generate isogenic revertant cells is a promising strategy for genome repair, and opens up new avenues for drug discovery13,14. However, the repair process remains problematic, and a precise and convenient genome editing procedure has not yet been developed.
Artificial genome repair and/or modification generally starts from a target-specific double-strand break generated by site-specific nucleases15,16. Double-strand breaks are then repaired by a cell’s own genome repair machineries15,16. However, most of the break sites are incorrectly repaired by non-homologous end joining, and can result in gene knockout through the generation of non-specific insertions or deletions (indels)17,18. If a repair template carrying a repair base is co-administrated, however, the cleavage sites can be accurately repaired via homology-directed repair (HDR)15,16. Site-specific nucleases, such as transcription activator-like effector nucleases or the Streptococcus pyogenes (Sp) Cas9 nuclease, are typically used as genome editing tools for human iPSCs19,20,21.
SpCas9 is a type II-A endonuclease in the class 2 clustered regularly interspaced short palindromic repeats (CRISPR)-Cas system that has been repurposed as a programmable site-specific nuclease for genome engineering22,23,24,25. Indeed, SpCas9 has become a popular genome editing tool for genetically modifying human pluripotent stem cells19. Despite its efficient cleavage activity, wild-type SpCas9 has a low DNA repair rate using HDR following plasmid-based administration. It re-cuts the repaired site because the guide RNA has a 1–2-base mismatch tolerance during sequence recognition, leading to incorrect repair by non-homologous end joining17,18. To prevent re-cutting, a blocking mutation must be introduced into the seed sequence of the guide RNA or into the protospacer-adjacent motif (PAM)26,27,28,29,30,31.
However, for “scarless” genome editing repairs with wild-type SpCas927,49, each method has its obvious strengths and weaknesses. For example, CORRECT, which includes excellent tricks to prevent re-cutting of the edited target by the editing tool, can be performed even if the target recognition ability of the genome editing tool is insufficient; however, the need for two consecutive editing steps27. In comparison, MhAX has high genome editing efficiency but cannot achieve completely scarless editing because single-base markers are required. Further, as with the CORRECT method, MhAX editing requires two consecutive edits49, increasing cost and time requirements.
Another recently-identified bacterial programmable site-specific nuclease, CRISPR-Cas12a/Cpf1, is a type V-A endonuclease belonging to the class 2 CRISPR-Cas system32. Among identified Cas12a enzymes, those from Acidaminococcus sp. BV3L6 (As) and Lachnospiraceae bacterium ND2006 show strong cleavage activity in mammalian cells32. These Cas12a endonucleases have unique features that complement Cas9, and expand the genome editing range. First, Cas12a recognises a T-rich PAM upstream of the protospacer, whereas Cas9 recognises a G-rich PAM downstream of the protospacer32. Second, two PAM-interacting variants have been generated that expand the Cas12a target range33,34. Third, Cas12a-mediated cleavage generates a staggered cut on the PAM-distal region of the target sequence, as opposed to the PAM-proximal blunt ends generated by Cas932. Finally, and perhaps most importantly, the guide or CRISPR RNA (crRNA) exhibits high-fidelity target recognition, meaning that Cas12a can precisely distinguish the target sequence at a single-base resolution35,36. Consequently, the resulting off-target effects are kept to a background level. These features suggest that Cas12a might be suitable for precise, disease-specific iPSC repair because its re-cut activity is low.
Cas12a has already been used to knock out pathogenic genes in cancer cells37, generate insertions or two-nucleotide substitutions in iPSCs38 and to induce exon-skipping in disease-specific iPSCs39. Thus, we investigated whether Cas12a could be used to carry out allele-specific single-nucleotide repair of iPSCs carrying the pathogenic SNVs found in MEN2B and DEB patients in a single round of genome editing. To accomplish this, we used an AsCas12a PAM variant and single-nucleotide mismatch detection polymerase chain reaction (SNMD-PCR) analysis in disease-specific iPSCs to develop a precise, convenient genome-editing procedure we have called One-SHOT (One allele-specific, single HDR and single-stranded oligodeoxynucleotide (ssODN), transient drug selection with SNMD-PCR screening). One-SHOT provides scarless single-nucleotide substitution of a pathogenic SNV in disease-specific iPSCs within 3 weeks. The final modification rate is within a practical range for hand-picking cloning. Our findings suggest that this simple, low cost procedure could be used for genome editing in a single step, drastically reducing the time currently needed for scarless SNV repair.
Results
Principles of One-SHOT repair of single-nucleotide mutations
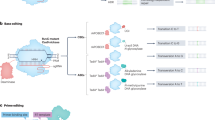
AsCas12a is a high-fidelity RNA-guided site-specific nuclease that binds to the target genomic DNA site via a 20-nt guide sequence in the crRNA, allowing it to discriminate the target sequence at the single nucleotide level (Fig. 1). Following the addition of a crRNA designed for a specific target sequence containing a single-nucleotide mutation, AsCas12a selectively binds to the target sequence on the mutant allele and induces a double-strand break, leaving the wild-type sequence on the alternative allele unaffected (Fig. 1). In the presence of a ssODN wild-type sequence template, the mutant nucleotide in the target sequence can be “repaired” to the wild-type sequence via the cellular HDR machinery. To mark the repaired allele, we labelled the ssODN with a single-nucleotide marker in the vicinity of the mutant nucleotide. This label allowed us to easily identify the gene-repaired clones by allele-specific amplification40,41,42/SNMD-PCR detection of the single-nucleotide marker (Fig. 1). A complete outline of the One-SHOT workflow for SNV repair is provided in the Supplementary Information and in Supplementary Fig. S1.

One-SHOT principles. AsCas12a (pale yellow) and crRNA (orange and grey lines) selectively bind to a target sequence containing a pathogenic SNV (red triangle) on the target allele. Binding leads to a double-strand break in the target sequence on the target allele (left) but not in the corresponding wild-type sequence containing the wild-type nucleotide (blue triangle) on the alternative, non-target (wild-type) allele (right). When the ssODN repair template (blue-green line) with the wild-type nucleotide (blue triangle) and a single-nucleotide marker (a silent mutation for SNMD-PCR screening, green triangle) is co-transfected with AsCas12a into the cells, the target site on the pathogenic allele is repaired using the template by the endogenous HDR machinery. In this case, the intended gene-edited clones are easily identified by positive screening for the single-nucleotide marker because the repaired, ex-pathogenic allele now carries the single-nucleotide marker.
Allele-specific single-nucleotide substitution in MEN2B-specific iPSCs
Before carrying out allele-specific single-nucleotide repair of the pathogenic RET mutation, we assessed whether the One-SHOT approach could be used to accomplish allele-specific single-nucleotide substitution of the wild-type allele.
We established FB4-14 human iPSCs from a patient with MEN2B using a Sendai viral vector protocol43. We then confirmed that the FB4-14 cells exhibited an embryonic stem cell-like morphology and expressed pluripotent gene markers, indicating that they were authentic iPSCs (Supplementary Fig. S2 and X7). To identify possible target sites for AsCas12a around the SNV of interest, we first searched for PAMs recognised by wild-type AsCas12a or the RR and RVR variants, which recognise TYCV and TATV PAMs, respectively33,34. We identified two PAM sites for the RR variant (TYCV, Y = C/T, V = A/C/G): TTCC, located 12-bp upstream of the target nucleotide on the sense strand, and TTCA, located 7-bp upstream of the target nucleotide on the antisense strand (Fig. 2a, magenta lines). Based on this information, we designed two pairs of crRNAs: crRNA_RET-1 + and crRNA_RET-1 m, and crRNA_RET-2 + and crRNA_RET-2 m, which contain guide sequences that specifically recognise wild-type and mutant target sequences, respectively (Fig. 2a).

Single-nucleotide substitution of the RET wild-type sequence in MEN2B iPSCs. (a) Human RET locus containing the MEN2B mutation and crRNA of AsCas12a_RR for the mutation. Top: exon 16 of the RET locus. Middle: the wild-type (WT) allele sequence. Blue letters indicate the wild-type nucleotide at Met918 (underlined). Bottom: the mutant allele sequence. Red letters indicate the single missense mutation caused by a T>C substitution, producing a Met918Thr substitution (underlined). Coloured lines indicate the guide sequence template for the crRNA. The pink line indicates the AsCas12a_RR PAM. Coloured dashed lines indicate the sites cleaved by AsCas12a_RR with the corresponding crRNA. (b) T7E1 assay using human wild-type iPSCs (409B2) electroporated with AsCas12a_RR and the different crRNAs (crRNA_RET-1 + , crRNA_RET-1 m, crRNA_RET-2 + or crRNA_RET-2 m) targeting exon 16. Left: the cropped gel images. Arrowheads indicate cleaved bands. The full-length gels are presented in Supplementary Figure S7. Right: statistical analysis of the cleavage activity and specificity of AsCas12a_RR with the crRNAs following selection with different concentrations of puromycin. (c) HDR-mediated editing for generating artificial homozygous MEN2B using AsCas12a_RR with crRNA_RET-1 + selectively targeting RET_Met918 in the wild-type allele. (d) SNMD-PCR analysis of the first round of screening. The cropped gel image is shown here. The arrowhead indicates positive PCR amplicon (202 bp). The full-length gel is presented in Supplementary Figure S8. (e) Sequencing of the original and modified MEN2B iPSCs (FB4-14). Top: original sequence of RET exon 16 with a T > C substitution in the MEN2B mutant allele. Bottom: the modified RET sequence. The T > C substitution resulting in a homozygous Met > Thr substitution. Red and blue arrows indicate the positions of the pathogenic SNV and the single-nucleotide marker, respectively. Underlining indicates the codons affected by the editing. A more detailed explanation is provided in the “Extended Figure Legends” in the Supplementary Information.
To test the cleavage activity and target-recognition specificity of AsCas12a_RR using these crRNAs, we performed a T7E1 assay using 409B2 human iPSCs carrying the wild-type RET sequence in the target site (Fig. 2a, middle). The crRNAs for the wild-type sequence (crRNA_RET-1 + and crRNA_RET-2 +) each exhibited significant cleavage activity towards the wild-type target sequence (Fig. 2b; P < 0.0001 and P = 0.0246, respectively). By contrast, the crRNAs for the mutant sequence (crRNA_RET-1 m and crRNA_RET-2 m) showed extremely weak activity (Fig. 2b; P > 0.9999 for both). A more accurate ICE analysis showed no significant activity of the crRNAs on the WT allele (Supplementary Fig. X2a). These results indicate that the crRNAs for the mutant sequence do not have significant, if any, activity on the WT allele”.
However, the observed cleavage activity of AsCas12a_RR in conjunction with crRNA_RET-1 + was significantly higher than that with crRNA_RET-2 + (Fig. 2b; P < 0.0001, Supplementary Fig. X2a). Puromycin treatment further promoted the cleavage activity of AsCas12a_RR with crRNA_RET-1 + (Fig. 2b; P < 0.0001).
To test the applicability of the method to carry out allele-specific single-nucleotide substitution in human iPSCs, we attempted to replace the wild-type nucleotide (RET: c.2753T) at the Met918 site in the wild-type allele in FB4-14 MEN2B-iPSCs (Fig. 2c). Following electroporation of the pY211-puro vector, which expresses AsCas12a_RR and crRNA_RET-1 + (Fig. 2c. blue line), and a ssODN modification template (ssODN_RET_M918T_I913silentC) carrying both a variant nucleotide at Met918 and a single-nucleotide marker at Ile913 (Fig. 2c, red C and light green C, respectively) into FB4-14 cells, we conducted SNMD-PCR screening. Overall, 12/384 clones were positive for the substitution (Fig. 2c, d and GE1 in Table 1). Direct sequencing of the target sequence revealed that 7/12 clones contained the wild-type allele-specific introduction of the mutant nucleotide at the target site (T > C substitution resulting in the Met918Thr substitution; Fig. 2e, red arrow), along with the single-nucleotide marker (T > C substitution leading to a silent mutation at Ile913; Fig. 2e, blue arrow). The HDR efficiency was 1.8% (Table 1).
We then searched for off-target sequences corresponding to the target sequence using the web tool CHOPCHOP v244 and detected no indels in either of the predicted two off-target sites by Sanger sequencing (Table 2, GE1) and by AmpliSeq (Supplementary Table X4). These results indicated that the One-SHOT method could be used to replace a single nucleotide in an allele-specific manner while minimising off-target effects. As in the preliminary experiment, direct sequencing analysis around the target sites revealed no duplication events in the unintended gene-edited clones, suggesting that most of the intended gene-edited clones had clonally proliferated (Supplementary Fig. S3, GE1).
Allele-specific single-nucleotide repair of a pathogenic RET variant
To repair the pathogenic SNV (RET: c.2753T>C) in the mutant allele in FB4-14 cells, we first tested the cleavage activity and target recognition specificity of AsCas12a_RR using crRNA_RET-1 m and crRNA_RET-2 m (Fig. 2a) in a homozygous MEN2B iPSC line with mutations in RET exon 16 in both alleles (GE1-9, genotype: RETMet918Thr/Met918Thr; RETIle913 silentC/+) (Fig. 2e, bottom). The T7E1 assay confirmed that the MEN2B target sequence was selectively cleaved by AsCas12a_RR with either crRNA_RET-1 m or crRNA_RET-2 m, but not with crRNA_RET-1 + or crRNA_RET-2 + (Fig. 3a). The ICE analysis revealed that only the AsCas12a_RR with crRNA_RET-1 m exhibited strong cleavage activity on the target sequence (Supplementary Fig. X2b), therefore we selected the crRNA_RET-1 m for use in subsequent experiments.

Single-nucleotide substitution in the human mutant allele in MEN2B iPSCs. (a) T7E1 assay in human artificial homozygous MEN2B iPSCs (FB4-14 homologous cells, generated using the scheme shown in Fig. 2) electroporated with the pY211-puro expression vector, which expresses AsCas12a_RR plus crRNAs. Left: The cropped gel image of the T7E1 assay. AsCas12a_RR with crRNA_RET-1 m and crRNA_RET-2 m, designed to target the mutant allele, exhibited significant cleavage activity on the mutant allele. Arrowheads indicate cleaved bands. M marker, NC negative control. The full-length gel is presented in Supplementary Figure S9. Right: statistical analysis of the cleavage activity and specificity. Bars indicate the mean and S.E.M. from experiments performed in biological triplicate. Data were analysed by one-way ANOVA with Tukey’s multiple comparison test. ns not significant. (b) HDR-mediated editing for repair of the MEN2B genome. The light green line indicates the target of crRNA_RET-1 m. The pink line indicates the AsCas12a_RR PAM. Red, blue and light green letters denote the pathogenic SNV, the wild-type nucleotide and the single-nucleotide marker, respectively. The pale blue and green columns indicate the positions of the pathogenic SNV and the single-nucleotide marker, respectively. The arrow indicates the position of the forward primer for the SNMD-PCR. (c) First round of SNMD-PCR screening. The arrowhead indicates the expected amplicon (202 bp). The full-length gel is presented in Supplementary Figure S10. (d) Sequencing chromatogram for the original and repaired MEN2B iPSCs. Top: the original sequence of RET exon 16 with a T>C substitution in the mutant allele. Bottom: the repaired RET sequence. Red and blue arrows indicate the positions of the pathogenic SNV and the single-nucleotide marker, respectively. Underlining indicates the codons affected by the editing.
We then carried out One-SHOT repair in FB4-14 cells using AsCas12a_RR with crRNA1m and a ssODN repair template containing a repair nucleotide at Met918 and a single-nucleotide marker at Ile913 (Fig. 3b, T in blue and C in light green, respectively). Subsequent SNMD-PCR screening showed that 17/344 clones were positive (Fig. 3c, GE2 in Table 1), while direct sequencing confirmed that 11 of the positive clones contained the introduced wild-type nucleotide at the target site (C > T substitution), leading to a Thr918Met substitution (repair) (Fig. 3d, red arrow). These clones also contained the single-nucleotide marker (T > C substitution), leading to a silent mutation at Ile913 (Fig. 3d, blue arrow). The overall HDR efficiency was 3.2% (Table 1, GE2), and we detected no off-target effects by Sanger sequencing (Table 2, GE2) and by AmpliSeq (Supplementary Table X4). As in the preliminary experiment, direct sequencing analysis around the target sites revealed no duplication events in the unintended gene-edited clones, suggesting that most of the intended gene-edited clones had clonally proliferated (Supplementary Fig. S3, GE2).
Allele-specific single nucleotide repair of a pathogenic variants in RET and COL7A1 without a single-nucleotide marker
We next investigated whether the One-SHOT method could be used to repair the pathogenic SNV in RET without including the single-nucleotide marker, which would achieve true scarless repair. We therefore performed One-SHOT repair in the FB4-14 cells using AsCas12a_RR, crRNA_RET-1 m and the ssODN repair template with only a wild-type nucleotide at Met918. In the subsequent SNMD-PCR screening for the pathogenic SNV, no amplicons were obtained from repaired clones because the pathogenic SNV was lost from the mutant allele (Fig. 4a). Overall, we identified 44 negative clones by SNMD-PCR screening for the pathogenic SNV, and direct sequencing revealed that 5/44 carried only the wild-type nucleotide at Met918 (Fig. 4c,d and GE4 in Table 1). In this experiment, the overall HDR efficiency was 2.0% (Table 1, GE4), and no indels were detected in the two predicted off-target sites by Sanger sequencing (Table 2, GE4) and by AmpliSeq (Supplementary Table X4).

Repairing the pathogenic single-nucleotide variant (SNV) with scarless One-SHOT. (a) Scarless One-SHOT. The target site (left panel) on the pathogenic allele was repaired using the One-SHOT approach with a normal ssODN carrying no single-nucleotide markers. In this case, the intended gene-edited clones were identified by negative screening for the pathogenic SNV because the repaired, ex-pathogenic allele had lost the pathogenic SNV (right panel). Red and blue triangles indicate the pathogenic SNV and the wild-type nucleotide, respectively. (b) HDR-mediated scarless editing repair of the MEN2B genome using AsCas12a_RR with crRNA_RET-1 m selectively targeting the RET_Met918Thr site in the mutant allele (see Fig. 3b for the strategy). The PCR primer position is indicated by the black arrow. (c) Negative screening by SNMD-PCR as the first round of screening. The arrowheads denote the intended clones from this screening (188 bp). The full-length gel is presented in Supplementary Figure S11. (d) Sequencing chromatograms for the original and repaired MEN2B iPSCs. Top: the original sequence of RET exon 16 with a T>C substitution in the MEN2B mutant allele. Bottom: the repaired sequence of RET exon 16 (C>T substitution). Red arrows indicate the position of the pathogenic SNV. Underlining indicates the codons affected by the editing.
We next attempted to perform scarless repair of a pathogenic SNV in iPSCs derived from a patient with DEB to confirm the applicability of the approach for other hereditary diseases. We generated iPSCs from a patient with DEB (autosomal recessive compound mutation: COL7A1p.Gly2138Ter/+; COL7A1c.3591del13insGG/+) and aimed to substitute the pathogenic SNV (c.6412G > T (p.Gly2138Ter)) in exon 78 (Supplementary Fig. S4a,b). Scarless One-SHOT using AsCas12a_RR with crRNA_COL7A1-1 m plus the repair template scarlessly repaired the pathogenic SNV in the mutant allele (Supplementary Fig. S4c,d,e and GE5 in Table 1), with a substitution rate of 7.5%. No indels were detected in the seven predicted off-target sites (Supplementary Table S2, Supplementary Table X4). Unlike the scarless One-SHOT for RET_Met918Thr in FB4-14 cells (Fig. 4, GE4 in Table 1), identical sequences (~ 22%) within the target site were observed among the unintended gene-edited clones, suggesting that these clones were likely duplicated (Supplementary Fig. S3, GE5).
Discussion
Many hereditary human diseases are caused by single-nucleotide mutations. These single-base alterations have the potential to drastically alter protein structure and function. Although most single-nucleotide mutations are completely harmless (silent), repair of pathogenic SNVs would significantly improve the quality of life and life expectancy of patients with hereditary diseases. Thus, in the present study, we investigated whether we could achieve scarless repair of pathogenic SNVs in pluripotent stem cells from patients with two different types of hereditary disease: MEN2B and DEB. More importantly, we aimed to carry out the repairs in a single step.
Using the One-SHOT approach developed in this study, we successfully repaired a RET gene SNV in MEN2B iPSCs with the addition of a single-nucleotide selective marker in a single step. We then confirmed that the same technique could be used to carry out scarless repair in MEN2B- and DEB-specific iPSCs without the need for the single-nucleotide marker. Scarless repair, where no trace of gene editing is left around the target sequence, is the goal of any gene editing technique because it safely repairs mutations in non-coding genomic regions without any secondary effects. In contrast, the inclusion of marker sequences during gene editing can have downstream effects. Such secondary effects include the introduction of non-coding SNVs to cryptic splice sites, causing abnormal RNA splicing45,46, and mutations that introduce a premature termination codon, resulting in unstable mRNA46. Non-coding mutations affecting regulatory elements can also interfere with gene regulation through loss of function, resulting in reduced gene expression, or gain of function, resulting in gene mis- or overexpression47,48. Therefore, scarless repair is crucial for maintaining genome integrity and preventing unknown secondary effects in the target gene.
Several other methods of pathogenic SNV repair have been developed, including CORRECT26,27 and MhAX49. However, all currently available methods have inherent obstacles to achieving scarless SNV repair in a fast and error-free manner. To overcome some of these obstacles, we used the AsCas12a nuclease, which has high-fidelity target-recognition35,36, circumventing the need for a blocking base to inhibit re-cutting, as is required in other methods26,27. We also performed SNMD-PCR-based negative screening for the pathogenic SNV, which easily detects candidate clones containing the intended alteration. As a result of these modifications, we achieved absolute scarless editing of the RET and COL7A1 SNVs (see Fig. 4, Supplementary Fig. S4 and GE4 and GE5 in Table 1). Another advantage of the AsCas12a nuclease was the ability to carry out SNV repair in a single step because only one round of HDR is required for gene editing (Fig. 1). The One-SHOT method was used to repair the SNVs in RET and COL7A1 within a 3-week period with sufficient efficiency for hand-picking. In contrast, other methods can take up to 2–3 months to generate the intended gene-edited clone because two rounds of HDR/MMEJ may be required26,27,49. However, similar to our approach, the CORRECT method can achieve scarless single-nucleotide substitution, thus ensuring high sequence fidelity around the target site in gene-edited cells (Fig. 4 and Supplementary Fig. S4). Conversely, MhAX leaves a silent single nucleotide mutation around the target site for use in screening49. Another difference is that the dsDNA template in MhAX can be randomly integrated into the genome (outside of the target site) by non-homologous end joining50, whereas the ssODN templates used for One-SHOT/scarless-One-SHOT and CORRECT approaches are not randomly integrated51. Thus, the One-SHOT method developed for SNV repair in the current study appears to have several advantages over currently available methods (see Supplementary Table S3).
In the CORRECT procedure, the cut-to-mutation distance (the distance between the CRISPR-SpCas9 cleavage site and the blocking mutation) is a crucial factor for HDR efficiency and zygosity determination26,27. We therefore searched for more appropriate sites for the single-nucleotide markers by first comparing the efficacies of three single-nucleotide markers set in different positions around the target site using a PCR-restriction fragment length polymorphism (RFLP) assay52. We found that two of the markers showed similar HDR-specific cleavage activity, while no cleavage activity was detected for the third marker (Supplementary Information and Supplementary Fig. S5), suggesting that Ile920 could be used as an alternative single-nucleotide marker. Testing of HDR efficiency in FB4-14 cells following One-SHOT repair using the alternative marker again confirmed that the single-nucleotide substitutions in the gene-edited clones were effectively detected by positive screening using SNMD-PCR for a single-nucleotide marker (Supplementary Information and Supplementary Fig. S5). We do note, however, that the efficiency of identification might depend on the position of the single-nucleotide marker and the primers used for SNMD-PCR.
Despite our success in repairing the pathogenic SNVs in a single step, the study has several limitations. The One-SHOT method only requires one PCR run, thereby reducing the time and cost compared with standard PCR–RFLP screening-based methods, which require up to three steps52,53. However, we found that false-positive clones are included in the population after the first SNMD-PCR screen (Supplementary Fig. X8). Therefore, we are currently designing a simple way to discriminate false clones from authentic clones using a PCR-based procedure. We also noted that the gene-edited cell lines generated by One-SHOT are not always clonal. This situation arises because high cell densities occur in the culture during puromycin selection (2 days) and in the recovery culture (1–2 days) prior to clonal expansion. However, assessment of our data suggests that a 1-day recovery culture and sufficient single-cell suspension at the reseeding stage can prevent duplication and ensure clonal establishment of the gene-edited cells. Using the current protocol, we estimate that the HDR substitution rate is 1.8–7.5%. While this is sufficient to permit a hand-picking cloning protocol, it is lower than that achieved by Cas12a in fertilised eggs from model animals39,54 We hoped to improve this rate by combining One-SHOT with other procedures based on alternative principles, such as introducing a blocking base into the repair template26,27 and/or using HDR/NHEJ modification compounds38,63,64,65,66,67. We have examined whether the modification compounds can promote HDR, however, the compounds examined in this study had no HDR-promoting effects in our experimental system (Supplementary Fig. X4).
It is important to emphasise, though, that the procedure depends on high-fidelity target recognition by the site-specific nuclease. Thus, the only enzymes appropriate for the One-SHOT procedure include high-fidelity variants of engineered SpCas955,56,57,58 or naturally high-fidelity Cas9 orthologues59,60,61,62. Finally, while we confirmed the expression of pluripotency markers in the gene-edited clones (data not shown), we next aim to carry out functional analyses to confirm the differentiation potential of the repaired cells. Therefore, further work is needed to fine-tune the protocol and to confirm differentiation potential and functionality of the proteins in the corrected cell populations.
To increase the reliability of the One-SHOT method, it is important to show the robustness of One-SHOT and the fidelity of the repair. In order to demonstrate these issues, we performed targeted NGS-based deep AmpliSeq analysis of the target sequence. With regard to repair fidelity, the AmpliSeq analysis showed that accurate single-nucleotide substitutions were achieved by HDR that were faithful to the ssODN template and occurred at sufficient frequency (Supplementary Fig. X3a-c, 2.45–5.44%). These results suggest that the method has good repair fidelity. With regard to the robustness of One-SHOT method, the AmpliSeq analysis confirmed that single-nucleotide substitutions were reliably performed under various conditions (different targets, different templates), (Supplementary Fig. X3, Supplementary Table X4), suggesting that our genome editing method is robust.
Quality control for the gene-edited cells is quite important for research and industrial use. Here, we performed four tests (clonal purity, on-/off-target effects, karyotyping, and random integration of the plasmid vector) as a form of quality control for the gene-edited cells generated via the One-SHOT method. Our results confirmed the clonal purity of the cells (Supplementary Fig. X1) and showed that off-target effects were negligible (Supplementary Fig. X3, Supplementary Table X4). However, karyotyping (Supplementary Fig. X5) and plasmid integration analysis (Supplementary Fig. X6) uncovered low-frequency chromosomal abnormalities and low-frequency plasmid integration events. Analysis of the literature suggests that these are not problems unique to the One-SHOT method, and are also observed in other genome editing processes70,71. These findings suggest that strict quality control is required when creating and using gene-edited cells, depending on the purpose (basic research, drug discovery platform, cell transplantation).
In summary, repair of pathogenic SNVs causing hereditary diseases in humans using site-specific nucleases has been relatively difficult to date because of insufficient target recognition accuracy by wild-type SpCas9. By using AsCas12a as the genome editing tool, we successfully achieved repair of two different pathogenic SNVs in a single step. The One-SHOT procedure developed in this study is simple, fast, low-cost, efficient, and broadly applicable to genome-editing applications in basic biological and biomedical research. We anticipate that One-SHOT will become a powerful procedure for generating isogenic cells from disease-specific iPSCs and for repairing genomic mutations in somatic cells, germ-line stem cells, and iPSCs.
Materials and methods
FB4-14 and B117-3 iPSC clones
FB4-14 cells (MEN2B-specific iPSCs) were used in GE experiments 1–4 (GE1–4). The cells were generated from a 40-year-old male patient who carried an autosomal dominant mutation at the MEN2B disease locus. The disease is caused by a mono-allelic SNV (RET: c.2753T>C) in the tyrosine kinase domain of the RET gene at codon 918 in exon 16, resulting in a Met918Thr substitution7. In GE5, B117-3 cells (DEB-specific iPSCs) were used. These were generated from a 3-year-old male patient who carried DEB autosomal recessive compound mutations within the COL7A1 gene. These mutations consisted of a mono-allelic SNV (COL7A1: c.6412G > T) at codon 2,138 in exon 78, causing a nonsense mutation (p.Gly2138Ter), along with a mono-allelic indel (c.3591del 13insGG) resulting in a frame-shift in exon 27. For the initial T7E1 assay, wild-type iPSCs (409B2; RIKEN BRC, Japan; clone number HPS0076) were used. iPSC clones FB4-14 and B117-3 exhibited typical iPSC properties (Supplementary Figs. S2, S4a, and X7).
Transfection of iPSCs
iPSCs were electroporated with a specific pY211-puro vector and ssODN. Briefly, 1 × 106 cells were resuspended in 100 µl of OptiMEM (Thermo Fisher Scientific, USA) containing 10 µg of pY211-puro and 15 µg of ssODN (99 nucleotides, PAGE-purified; Sigma-Aldrich, USA). The cells were then electroporated (transfer pulse, 20 V; pulse length, 50 ms; number of pulses, 5) in a 2-mm gap-size cuvette using a Super Electroporator NEPA21 Type 2 instrument (NEPA GENE, Japan). Following electroporation, cells were transferred to Matrigel-Growth Factor Reduced (GFR)-coated 24-well plates containing mTeSR1 plus CloneR medium (STEMCELL Technologies, Canada) and cultured for 16 h. The cells were then cultured in mTeSR1 plus CloneR medium containing puromycin (0.5 µg/ml) for 48 h, and subsequently transferred back into mTeSR1 plus CloneR medium for 1–2 days to recover. After recovery, the cells were singularised using TrypLE (Gibco/Thermo Fisher Scientific, USA) and seeded at a low density (500–1,000 cells per 100 mm plate) in mTeSR1 plus Clone R medium for cloning.
Genotyping of single-cell-derived clones by SNMD-PCR and sequencing
To aid in the genotyping of single-cell-derived clones, ssODN templates for HDR were designed to introduce a single-nucleotide marker (a silent mutation) independent of the pathogenic SNV for SNMD-PCR screening. Genome-edited, single-cell-derived iPSC clones were cultured in master plates (Matrigel-GFR-coated 100-mm plates with 100 square grids (PetriSticker); Diversified Biotech Inc., USA) for 4 days in mTeSR1 plus CloneR medium, followed by 3 days in unsupplemented mTeSR1. The central portion of each colony (approximately 25–33%) was manually picked under a microscope68 and directly transferred to a PCR tube containing 10 µl of lysis buffer (DirectPCR Lysis Reagent Cell, Viagen Biotech, USA). The remaining colonies on the master plates were maintained for 2–3 days under the same conditions.
To identify the HDR-targeted clones by SNMD-PCR, the genomic regions surrounding the target loci were amplified using HiDi DNA polymerase69 (myPOLS Biotec GmbH, Germany) and the corresponding single-nucleotide marker-specific or allele-specific primer pair (Supplementary Table S1). Amplicons were analysed by 2% agarose gel electrophoresis. Gene Ladder Wide I (Nippon Gene, Japan) was used as DNA molecular size marker. The presence of the single-nucleotide marker introduced by HDR and the zygosity of the pathogenic variants in the clones were confirmed by Sanger sequencing. For these assays, 450–500-bp amplicons around the gene-edited locus were PCR-amplified using the specific primer pair and Tks Gflex DNA polymerase (Takara Bio, Japan). Sequencing was performed using a BigDye Terminator v3.1 Cycle Sequencing Kit (Thermo Fisher Scientific, USA).
One-SHOT procedure
To perform allele-specific single-nucleotide substitution at the pathogenic SNV in the human iPSCs, we developed the One-SHOT repair method using high-fidelity RNA-guided site-specific nuclease AsCas12a. To carry out the repair, 1 × 106 iPSCs were electroporated with a pY211-puro vector expressing AsCas12a_RR, crRNA corresponding to the target sequence and the puromycin resistance gene. In the positive-screening protocol for standard genome editing (standard One-SHOT), a ssODN was used to introduce the intended mutation (M) and a single-nucleotide marker (S) (the MS template). Cells transiently expressing AsCas12a-crRNA-puroR were treated with puromycin, and surviving cells were enzymatically singularised and plated at a low density (500–1,000 cells per 100-mm plate) to permit growth of the single-cell-derived clones. The presence of the single-nucleotide marker was detected by SNMD-PCR and confirmed by Sanger sequencing. In the case of the negative-screening protocol for scarless genome editing (scarless One-SHOT), a ssODN that only contained the intended mutation (the M template) was introduced. The presence of the M mutation was negatively detected by SNMD-PCR and confirmed by Sanger sequencing. The efficiency of introducing the intended mutation by One-SHOT was determined by Sanger sequencing of the established and expanded clones. A detailed explanation of each step is provided in the Supplementary Information (Extended Materials and Methods).
Statistical analyses
GraphPad Prism version 7 (GraphPad Software Inc., USA) was used for all statistical analyses. An analysis of variance (ANOVA) was performed on all data, followed by Tukey’s multiple comparisons test for the T7E1 assays and ICE analysis, or Sidak’s multiple comparisons test for the PCR–RFLP assay and Clonal Purity analysis.
Ethical statement
All experiments and methods were performed in accordance with relevant guidelines and regulations. All experimental protocols were approved by the Fukushima Medical University Institutional Review Board. All experimental procedures involving human samples were approved by the following ethics committees: the Human Genome and Gene Analysis Research Committee at Fukushima Medical University (approval number 2186), the Epidemiological and General Research Committee of the Faculty of Life Science, Kumamoto University, the Human Genome and Gene Analysis Research Committee of the Faculty of Life Sciences, Kumamoto University, and the Clinical Research and Advanced Medical Technology Committee, Kumamoto University (approval numbers 318, 153 and 1,018, respectively). Blood samples (10 ml) were collected from the two patients involved in this study, and T-cells were isolated using Ficoll-Paque PREMIUM density gradient media (GE Healthcare). Informed consent was obtained from the participant or the participant’s parents prior to the study.
Data availability
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
References
International HapMap Consortium. A haplotype map of the human genome. Nature 437, 1299–1320 (2005).
Katsonis, P. et al. Single nucleotide variations: Biological impact and theoretical interpretation. Protein Sci. 23, 1650–1666 (2014).
Huddleston, J. et al. Discovery and genotyping of structural variation from long-read haploid genome sequence data. Genome Res. 27, 677–685 (2017).
Ott, J., Kamatani, Y. & Lathrop, M. Family-based designs for genome-wide association studies. Nat. Rev. Genet. 12, 465–474 (2011).
Ozaki, K. et al. Functional SNPs in the lymphotoxin-alpha gene that are associated with susceptibility to myocardial infarction. Nat. Genet. 32, 650–654 (2002).
Wellcome Trust Case Control Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature 447, 661–678 (2007).
Wells, S. A. Jr. et al. Multiple endocrine neoplasia type 2 and familial medullary thyroid carcinoma: an update. J. Clin. Endocrinol. Metab. 98, 3149–3164 (2013).
Shinkuma, S. Dystrophic epidermolysis bullosa: A review. Clin. Cosmet. Investig. Dermatol. 8, 275–284 (2015).
Park, I. H. et al. Disease-specific induced pluripotent stem cells. Cell 134, 877–886 (2008).
Sanchez-Danes, A. et al. Disease-specific phenotypes in dopamine neurons from human iPS-based models of genetic and sporadic Parkinson’s disease. EMBO Mol. Med. 4, 380–395 (2012).
Soga, M. et al. HPGCD outperforms HPBCD as a potential treatment for Niemann-Pick disease type C during disease modeling with iPS cells. Stem Cells 33, 1075–1088 (2015).
Somers, A. et al. Generation of transgene-free lung disease-specific human induced pluripotent stem cells using a single excisable lentiviral stem cell cassette. Stem Cells 28, 1728–1740 (2010).
Peitz, M., Jungverdorben, J. & Brustle, O. Disease-specific iPS cell models in neuroscience. Curr. Mol. Med. 13, 832–841 (2013).
Soldner, F. & Jaenisch, R. Medicine. iPSC disease modeling. Science 338, 1155–1156 (2012).
Rouet, P., Smih, F. & Jasin, M. Introduction of double-strand breaks into the genome of mouse cells by expression of a rare-cutting endonuclease. Mol. Cell. Biol. 14, 8096–8106 (1994).
Rouet, P., Smih, F. & Jasin, M. Expression of a site-specific endonuclease stimulates homologous recombination in mammalian cells. Proc. Natl. Acad. Sci. USA 91, 6064–6068 (1994).
Hsu, P. D., Lander, E. S. & Zhang, F. Development and applications of CRISPR-Cas9 for genome engineering. Cell 157, 1262–1278 (2014).
Lieber, M. R. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu. Rev. Biochem. 79, 181–211 (2010).
Hockemeyer, D. & Jaenisch, R. Induced pluripotent stem cells meet genome editing. Cell Stem Cell 18, 573–586 (2016).
Hockemeyer, D. et al. Genetic engineering of human pluripotent cells using TALE nucleases. Nat. Biotechnol. 29, 731–734 (2011).
Sexton, A. N. et al. Genetic and molecular identification of three human TPP1 functions in telomerase action: Recruitment, activation, and homeostasis set point regulation. Genes Dev. 28, 1885–1899 (2014).
Jinek, M. et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821 (2012).
Jinek, M. et al. RNA-programmed genome editing in human cells. eLife 2, 471 (2013).
Ran, F. A. et al. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 8, 2281–2308 (2013).
Mali, P. et al. RNA-guided human genome engineering via Cas9. Science 339, 823–826 (2013).
Kwart, D. et al. Precise and efficient scarless genome editing in stem cells using CORRECT. Nat. Protoc. 12, 329–354 (2017).
Paquet, D. et al. Efficient introduction of specific homozygous and heterozygous mutations using CRISPR/Cas9. Nature 533, 125–129 (2016).
Hsu, P. D. et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 31, 827–832 (2013).
Jiang, W. et al. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat. Biotechnol. 31, 233–239 (2013).
Kuscu, C. et al. Genome-wide analysis reveals characteristics of off-target sites bound by the Cas9 endonuclease. Nat. Biotechnol. 32, 677–683 (2014).
Pattanayak, V. et al. High-throughput profiling of off-target DNA cleavage reveals RNA-programmed Cas9 nuclease specificity. Nat. Biotechnol. 31, 839–843 (2013).
Zetsche, B. et al. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell 163, 759–771 (2015).
Gao, L. et al. Engineered Cpf1 variants with altered PAM specificities. Nat. Biotechnol. 35, 789–792 (2017).
Nishimasu, H. et al. Structural basis for the altered PAM recognition by engineered CRISPR-Cpf1. Mol. Cell. 67, 139–147 (2017).
Kim, D. et al. Genome-wide analysis reveals specificities of Cpf1 endonucleases in human cells. Nat. Biotechnol. 34, 863–868 (2016).
Kleinstiver, B. P. et al. Genome-wide specificities of CRISPR-Cas Cpf1 nucleases in human cells. Nat. Biotechnol. 34, 869–874 (2016).
Yang, M. et al. Targeted disruption of V600E-mutant BRAF gene by CRISPR-Cpf1. Mol. Ther. Nucleic Acids 8, 450–458 (2017).
Ma, X. et al. Small molecules promote CRISPR-Cpf1-mediated genome editing in human pluripotent stem cells. Nat. Commun. 9, 1303 (2018).
Zhang, Y. et al. CRISPR-Cpf1 correction of muscular dystrophy mutations in human cardiomyocytes and mice. Sci. Adv. 3, e1602814 (2017).
Myakishev, M. V. et al. High-throughput SNP genotyping by allele-specific PCR with universal energy-transfer-labeled primers. Genome Res. 11, 163–169 (2001).
Pastinen, T. et al. A system for specific, high-throughput genotyping by allele-specific primer extension on microarrays. Genome Res. 10, 1031–1042 (2000).
Shi, M. M., Bleavins, M. R. & de la Iglesia, F. A. Technologies for detecting genetic polymorphisms in pharmacogenomics. Mol. Diagn. 4, 343–351 (1999).
Fujie, Y. et al. New type of Sendai virus vector provides transgene-free iPS cells derived from chimpanzee blood. PLoS ONE 9, e113052 (2014).
Labun, K. et al. CHOPCHOP v2: A web tool for the next generation of CRISPR genome engineering. Nucleic Acids Res. 44, W272-276 (2016).
Cooper, D. N., Krawczak, M. & Antonorakis, S. E. The nature and mechanisms of human gene mutation. In Metabolic and Molecular Basis of Inherited Disease (eds Scriver, C. et al.) 259–291 (McGraw-Hill, New York, 1995).
Strachan, T. & Read, A. P. Pathogenic mutations. In Human Molecular Genetics (eds Strachan, T. & Read, A. P.) 222–227 (BIOS Scientific Publishers Ltd, Oxford, 1999).
Zhang, F. & Lupski, J. R. Non-coding genetic variants in human disease. Hum. Mol. Genet. 24, R102–R110 (2015).
Spielmann, M. & Mundlos, S. Looking beyond the genes: the role of non-coding variants in human disease. Hum. Mol. Genet. 25, R157–R165 (2016).
Kim, S. I. et al. Microhomology-assisted scarless genome editing in human iPSCs. Nat. Commun. 9, 939 (2018).
Tsai, S. Q. et al. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat. Biotechnol. 33, 187–197 (2015).
Yoshimi, K. et al. ssODN-mediated knock-in with CRISPR-Cas for large genomic regions in zygotes. Nat. Commun. 7, 10431 (2016).
Nomura, N. et al. PCR-RFLP: A new HLA-DNA typing method tested in bone marrow transplantation. Transplant Proc. 23, 431–433 (1991).
Nomura, N. et al. HLA-DQB1 genotyping by a modified PCR-RFLP method combined with group-specific primers. Tissue Antigens 38, 53–59 (1991).
Moreno-Mateos, M. A. et al. CRISPR-Cpf1 mediates efficient homology-directed repair and temperature-controlled genome editing. Nat. Commun. 8, 2024 (2017).
Slaymaker, I. M. et al. Rationally engineered Cas9 nucleases with improved specificity. Science 351, 84–88 (2016).
Kleinstiver, B. P. et al. High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature 529, 490–495 (2016).
Chen, J. S. et al. Enhanced proofreading governs CRISPR-Cas9 targeting accuracy. Nature 550, 407–410 (2017).
Hu, J. H. et al. Evolved Cas9 variants with broad PAM compatibility and high DNA specificity. Nature 556, 57–63 (2018).
Kim, E. et al. In vivo genome editing with a small Cas9 orthologue derived from Campylobacter jejuni. Nat. Commun. 8, 14500 (2017).
Chen, F. et al. Targeted activation of diverse CRISPR-Cas systems for mammalian genome editing via proximal CRISPR targeting. Nat. Commun. 8, 14958 (2017).
Amrani, N. et al. NmeCas9 is an intrinsically high-fidelity genome-editing platform. Genome Biol. 19, 214 (2018).
Chatterjee, P., Jakimo, N. & Jacobson, J. M. Minimal PAM specificity of a highly similar SpCas9 ortholog. Sci. Adv. 4, 766 (2018).
Yu, C. et al. Small molecules enhance CRISPR genome editing in pluripotent stem cells. Cell Stem Cell 16, 142–147 (2015).
Lin, S. et al. Nocodazole Enhanced homology-directed human genome engineering by controlled timing of CRISPR/Cas9 delivery. eLife 3, e04766 (2014).
Chu, V. T. et al. Increasing the efficiency of homology-directed repair for CRISPR-Cas9-induced precise gene editing in mammalian cells. Nat. Biotechnol. 33, 543–548 (2015).
Maruyama, T. et al. Increasing the efficiency of precise genome editing with CRISPR-Cas9 by inhibition of nonhomologous end joining. Nat. Biotechnol. 33, 538–542 (2015).
Robert, F. et al. Pharmacological inhibition of DNA-PK stimulates Cas9-mediated genome editing. Genome Med. 7, 93 (2015).
Li, H. L. et al. Efficient genomic correction methods in human iPS cells using CRISPR-Cas9 system. Methods 101, 27–35 (2016).
Drum, M. et al. Variants of a Thermus aquaticus DNA polymerase with increased selectivity for applications in allele- and methylation-specific amplification. PLoS ONE 9, e96640 (2014).
Kim, S. et al. Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Res. 24, 1012–1019 (2014).
Cullot, G. et al. CRISPR-Cas9 genome editing induces megabase-scale chromosomal truncations. Nat. Commun. 10, 1136 (2019).
Acknowledgements
This work was supported in part by donations and collaborative research grants (project numbers KI26901, KY29002) from Zenoaq Resource Co., Ltd., the Ministry of Health, Labour, and Welfare of Japan, the Japan Agency for Medical Research and Development (A-MED; to T. E.), and from JSPS KAKENHI (Grant number 16K10546; to I.N. and Y.Y., 19K09030; to Sa. S. and Y.Y.). The authors thank Dr Feng Zhang for providing pY211, Dr Dorus Gadella for providing pSGFP2-C1, and Dr Frank Sinicrope for providing pSIH-H1-Puro. We also thank Prof. Kohno and staff at the Department of Gastrointestinal Tract Surgery, Fukushima Medical University (FMU), for providing general equipment, Prof. Sekine and Dr Machida (Department of Immunology, FMU) for cell sorting and plate reading, Prof. Wada and Ms. Takeuchi (Department of Cell Science, Institute of Biomedical Sciences, FMU) for DNA sequencing, and Dr Kariya (Department of Biochemistry, FMU) for use of a NanoDrop 2000c. We thank Tamsin Sheen, PhD, and Sandra Cheesman, PhD, from Edanz Group (www.edanzediting.com/ac) for editing a draft of this manuscript.
Author information
Authors and Affiliations
Contributions
Y.Y. and T.E. conceived and designed the study, Y.Y., N.O., K.Y., S.N., Y.S., and K.I. collected and/or assembled data, Y.Y. analysed and interpreted data, N.O., K.Y., S.N., and Y.S. provided administrative support, Y.Y., Sh.S., I.N., S.T., Sa.S., and T.E. provided financial support, Sh.S., M.G., and I.N. provided study materials or patients, and Y.Y. and T.E. wrote the manuscript. All authors approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yokouchi, Y., Suzuki, S., Ohtsuki, N. et al. Rapid repair of human disease-specific single-nucleotide variants by One-SHOT genome editing. Sci Rep 10, 13927 (2020). https://doi.org/10.1038/s41598-020-70401-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-70401-7
This article is cited by
-
C and G are frequently mutated into T and A in coding regions of human genes
Molecular Genetics and Genomics (2024)
-
Nucleotid-genaue PCR: Von der Mutationsdetektion zur Genom-Editierung
BIOspektrum (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
