

















« Prev Next »
Sickle-cell anemia (SCA) is a disease that links biochemistry, pathology, natural selection, population genetics, gene expression, and genomics. Although the disease has existed for thousands of years, it wasn't until 1910 that the symptom complex featuring anemia, recurrent fevers, and bouts of horrific pain, often accompanied by sudden death, became a recognized clinical entity in Western medicine. In that year, James Herrick published a report of a dental student from Barbados with fever, recurrent pneumonia, and sickle-shaped red blood cells (RBCs) (Herrick, 1910). We now know that SCA is an autosomal recessive disorder in which patients inherit one mutated copy of the β-globin gene from each parent (Neel, 1949). The parents usually carry one wild-type and one sickle β-globin gene and are said to have sickle-cell trait, yet they have none of the disease phenotypes or consequences.
Biochemical Basis of Sickle-Cell Pathology
In 1949, Linus Pauling and associates dubbed SCA the first "molecular disease" and coined the termed "molecular medicine." They showed that hemoglobin from SCA patients had different physical properties than hemoglobin from wild-type individuals, whereas hemoglobin from individuals with sickle-cell trait had properties of both types of hemoglobin (Pauling et al., 1949). Adult hemoglobin consists of two α-globin chains and two β-globin chains (Rhinesmith et al., 1957; Rhinesmith et al., 1958). In the 1950s, Vernon Ingram demonstrated that the only structural difference between normal adult hemoglobin and sickle-cell hemoglobin is the replacement of glutamic acid with valine in the β-globin amino acid chain (Ingram, 1957; 1959). At the DNA level, this corresponds to a single base change, from adenine to thymine, within the sixth codon (Marotta et al., 1977). Loss of the negatively charged glutamic acid results in altered electrophoretic mobility.
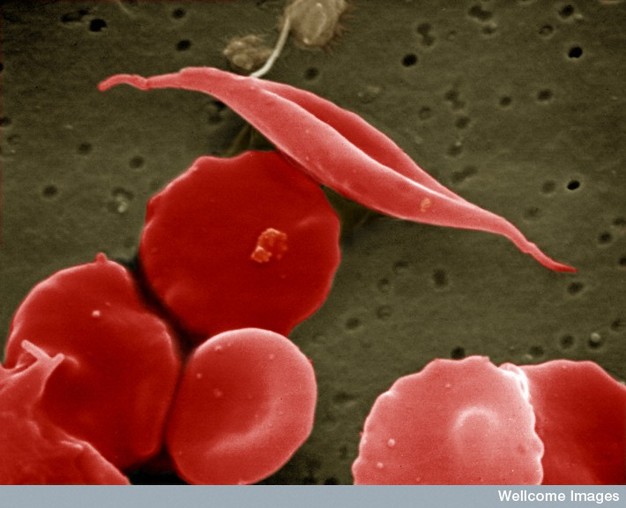
Figure 1: Sickle-cell anemia is characterized by deformed red blood cells.
A sickle-shaped red blood cell is shown among a group of healthy red blood cells. A change in a single amino acid in one of the hemoglobin proteins is responsible for causing the abnormal sickle shape of this red blood cell.
Creative Commons EM Unit, UCL Medical School, Royal Free Campus, Wellcome Images. 
In vivo, the consequences of this amino acid substitution only become apparent when oxygen dissociates from hemoglobin. Specifically, the deoxygenated hemoglobin molecule changes conformation such that the exposed valine sticks to a hydrophobic patch on a neighboring hemoglobin molecule. This rapidly leads to stacking of the hemoglobin into long polymers that deform the cell membrane into its characteristic sickle shape. Amazingly, the distorted RBCs resume a normal shape in the lungs when oxygen once again binds to hemoglobin. Over time, however, these transitions lead to irreversible distortions of the RBC membrane into a sickle shape (Figure 1) (Lux et al., 1976).
In the deoxygenated environment of tissue capillary beds, the misshapen RBCs are the cause of all the problems associated with SCA. The abnormally shaped cells can build up in capillaries and veins, obstructing (occluding) blood flow and leading to severe pain and tissue damage in almost any organ of the body. The cells' sickle shape and associated fragility result in rapid RBC destruction by a patient's own body. This leads to leads to anemia, as well as congestion and fibrosis of the spleen at an early age.
Mutant Beta-globin Leads to Positive Natural Selection
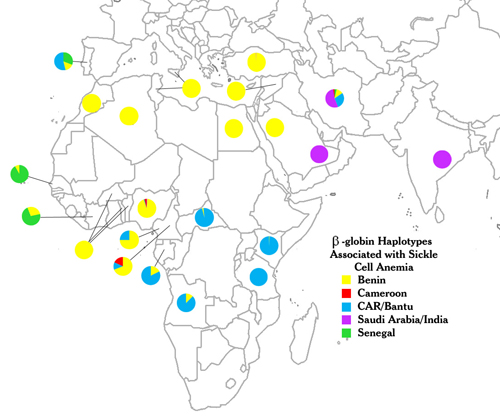
Figure 2: The distribution of sickle-cell anemia haplotypes among nations with high prevalence of the disease.
Five distinct β-globin haplotypes (indicated by colors) are found in patients with sickle-cell anemia. Each color represents a different haplotype named after the country in which it was first discovered, not necessarily its genetic origin. Indeed, these haplotypes are not restricted to the eponymous nation, and they can be found broadly distributed (i.e., Benin haplotype in multiple nations, or multiple haplotypes within a single nation). The haplotype data represented in the image were summarized from genetic epidemiological studies of sickle-cell patients across different regions. Because the number of patients per study and the population ascertainment methods are highly variable, the colors denote only the relative frequency of each haplotype within a given study group. CAR = Central African Republic. Data were taken from Monteiro et al., 1989; Nagel & Ranney, 1990; Oner et al., 1992; Rahimi et al., 2003; Schroeder et al., 1990; and references therein.
© 2010 Nature Education All rights reserved.
Allison and others used epidemiological, statistical, and experimental methods to prove an association between sickle-cell trait and malaria resistance (Allison, 2004). However, sickle-cell trait does not simply confer immunity to malaria to those who carry it. The protection it provides is relative, is specific to falciparum malaria (the most lethal form of the disease), is most apparent in regions where malaria poses a continuous (hyperendemic) threat, and is most important for children between the ages of two months and two years. Before two months, infants in hyperendemic environments are usually protected from malaria by acquired maternal antibodies. After about two years, most children who have survived their initial infection have developed protective antibodies that prevent the worst consequences of malaria. However, among children in the intermediate age window, a lower percentage of those with sickle-cell trait get cerebral malaria, severe malarial anemia, or other lethal consequences, and on average, the number of parasites in their bloodstream is decreased (Aidoo et al., 2002). These subtle differences mean that more individuals with the trait survive to reach reproductive age and pass on the mutant gene to their offspring. Still, despite much conjecture, researchers do not know exactly how the heterozygote state protects against malaria.
The Sickle-Cell Mutation Is Found in Many Populations
In 1978, Y. W. Kan and Andreé Dozy used the new tools of recombinant DNA and restriction enzymes to observe sequence differences (polymorphisms) in the DNA around the β-globin genes of different individuals. They went on to use these polymorphisms, reflected in the loss or gain of specific restriction enzyme cleavage sites, to trace the sickle β-globin allele within family pedigrees (Kan & Dozy, 1978a; 1978b; 1980). These studies were the first application of restriction fragment length polymorphisms (RFLPs) to follow disease genes, and they represented a milestone in human genetics. For SCA in particular, this effort paved the way for prenatal diagnosis and improved genetic counseling. In addition, it provided the tools used by other investigators to show that the mutation causing SCA arose multiple times during human history.
Unrelated individual humans differ from each other at the DNA level at approximately every 1,000 to 2,000 bases along each chromosome. Long segments of DNA that contain many such polymorphisms are termed haplotype blocks. Because polymorphisms are inherited, two individuals who share particular haplotype blocks also share a recent common ancestor. This property allows investigators to use haplotype analysis to identify relatedness among individuals.
Using RFLP analysis, geneticists found many distinct haplotypes in and around the tens of kilobases that make up the β-globin gene cluster. For instance, in 1984, Pagnier et al. reported that within four African populations with significant frequencies of sickle-cell trait, three distinct haplotypes were present among SCA patients that corresponded almost entirely to geographic origin. Patients from Benin and Algeria were homozygous for one haplotype (the Benin type), whereas more than 80% of patients from the Central African Republic (CAR) and from Senegal were homozygous for two other haplotypes (the Bantu and Senegal types, respectively). Although a few patients had two haplotypes resulting from population mixing, this study strongly suggested that the sickle-cell mutation arose independently at least three times in Africa and was selected for among geographically and reproductively isolated populations. A later study expanded the number of independent African initiating mutations to four, when Lapoumeroulie and colleagues identified a distinct haplotype among sickle-cell patients of the Eton ethnic group of eastern Cameroon (the Cameroon type, Figure 2) (Lapoumeroulie et al., 1992).
Not surprisingly, the haplotypes of African-American sickle-cell patients correspond to locations of forced African emigration to the New World through the African slave trade. In a study of 98 SCA patients from the state of Georgia, 54% had the Benin haplotype, 27% had the Bantu haplotype, and 15% had the Senegal haplotype (Hattori et al., 1986). Many of these patients were heterozygous for different haplotypes, reflecting the genetic mixing that has occurred in the New World. The historical spread of the sickle-cell mutation is also apparent among SCA patients of Greek, Portuguese, Sicilian, and other Southern European origins. In one well-documented study, SCA patients living in a formerly malarial region of central Portugal, and who were unaware of any African ancestry, were shown to have the same haplotypes around their β-globin clusters as West Africans. The African haplotypes corresponded to the geographic regions visited by Portuguese sailors in the 1400 through 1600s, implying the genetic mixing of Portuguese and West African populations and the subsequent incorporation of these offspring into the Portuguese population (Figure 2, top) (Monteiro et al., 1989).
In contrast to these non-African cases of SCA due to breeding between nations, investigators examining SCA patients in the eastern oases of Saudi Arabia and portions of central India discovered another distinct haplotype, indicating a fifth independent occurrence of the sickle-cell mutation in a region historically hyperendemic for malaria (Kulozik et al., 1986). Whether the geographic distribution of this mutation resulted from eastern migration to India or western migration to the Arabian peninsula is still unknown.
Does Haplotype Correlate with the Clinical Severity of SCA?
In an individual patient, the clinical severity of SCA can range from mild to severe. The degree of phenotypic variation in a disease caused by a (seemingly simple) single-base substitution illustrates the importance of other genetic loci in modifying disease severity. Recent anecdotal observations among the Arabian/Indian and Senegal haplotypes suggest they are associated with milder forms of SCA, whereas the Bantu haplotype is associated with a more severe clinical course (Steinberg, 2005). Interestingly, this observation correlates with differences in the mean levels of fetal hemoglobin (FH) in patients with different haplotypes. Individuals with the Arabian/Indian haplotype had an average 17% FH (Miller et al., 1987), those with the Senegal haplotype had an average 12.4% FH (Labie et al., 1985), and those with the Bantu and Benin haplotypes had even lower average FH. However, within each haplotype group, the range of FH levels is very broad.
What is the significance of FH to SCA disease mechanisms? FH has important clinical relevance. Unlike adult hemoglobin, FH consists of two α-globin chains and two γ-globin chains. In utero, FH is the major constituent of fetal RBCs, because β-globin is not expressed until the third trimester. Two nearly identical γ-globin genes are located next to the β-globin gene within the β-globin cluster. By birth, γ-globin production is normally switched off, but a significant fraction of RBCs still contain a mixture of fetal and adult hemoglobin. Because the sickle-cell mutation is present only on the β-globin gene, the problems of SCA only become apparent several months after birth, when the FH-containing RBCs have been diluted, worn out, and eliminated.
Patients with SCA tend to maintain perceptible levels of FH in their RBCs throughout childhood. This persistence has a protective effect on SCA severity, presumably by blocking the assembly of the long polymers of abnormal hemoglobin. Reactivating or increasing production of FH is the rationale for administering hydroxyurea, the only drug licensed to ameliorate the symptoms of SCA. By increasing FH content in the blood of some patients, hydroxyurea administration has been shown to decrease the incidence of painful episodes and serious vaso-occlusive incidents (Charache et al., 1995; Steinberg, 2005; Steinberg et al., 2003).
The correlation between sickle-cell haplotypes and FH levels has been extensively explored. Specific polymorphisms in the promoter regions of the nearby γ-globin genes are components of the Senegal and Arabian/Indian haplotypes, leading to the hypothesis that these sequence differences alter expression of the two γ-globin genes. However, there is so much phenotypic variation among individuals with a common haplotype that this hypothesis has proven unreliable despite the initial hope that haplotypes could be used as predictors of individual disease severity or guides to optimal treatment.
Genomics May Lead to a Better Understanding of SCA and Its Treatment
Unfortunately, knowing only the haplotype of an SCA patient may not be particularly helpful, as patients may experience symptoms of differing severity and varied responses to hydroxyurea regardless of haplotype. So, although these findings are of epidemiological and mechanistic interest, they do not influence the treatment or prognosis of a patient with SCA. Given the wide variability of disease severity, it is most likely that a patient's environment and other allelic differences within the β-gene cluster and unlinked loci all have significant influence on disease outcome.
Although environmental factors in sickle-cell disease have not been rigorously studied, the search for genetic modifiers of the disease has been ongoing. The most promising recent approach takes advantage of single nucleotide polymorphisms (SNPs) throughout the genome. DNA from individuals with or without a particular phenotype is analyzed for the presence or absence of SNPs. The frequency of any given SNP should be similar in both groups unless the SNP is located near a gene that influences the phenotype. By comparing the pattern of SNPs from a large number of cases and controls, multiple statistically significant loci can be detected.
Using a variety of phenotypes observed in subsets of SCA patients (including those with strokes, pulmonary hypertension, response to hydroxyurea, and osteonecrosis), a large number of candidate loci have been identified that may have predictive value (Rund & Fucharoen, 2008; Steinberg, 2008). More specific and validated results have come from studies examining which genomic loci affect the level of FH in both normal adults and patients with SCA. Using genome-wide association studies that simultaneously analyze hundreds of thousands of SNPs, the BCL11A locus on chromosome 2 and the HBS1L-MYB locus on chromosome 6 have been identified (Lettre et al., 2008; Sedgewick et al., 2008; Uda et al., 2008). Multiple SNP variants in these gene regions are associated with higher levels of FH and a milder course of disease. Together with the SNPs in the γ-globin region of the β-globin cluster, these loci account for more than 20% of the variance in FH levels among SCA patients in the United States and Brazil.
Summary
Although caused by a single amino acid substitution, SCA is a clinically heterogeneous disease for which multiple factors influence a particular patient's disease outcome. Since the discovery of SCA in 1910, analysis of this disorder has been on
the leading edge of biochemical and genetic science. Unfortunately, we still know little about how to predict a patient's clinical course. Large-scale genome-wide association studies of patients and controls are a promising new approach for helping researchers identify and analyze genetic loci that could lead to treatments tailored to specific patients based on their genotype, allowing for improved prognosis and quality of life. Thus, genetic analysis among SCA patients, though difficult to translate into immediate customization of treatment, may eventually lead to therapeutic improvements.
References and Recommended Reading
Aidoo, M., et al. Protective effects of the sickle-cell gene against malaria morbidity and mortality. Lancet 359, 1311–1312 (2002)
Allison, A. C. The distribution of the sickle-cell trait in East Africa and elsewhere, and its apparent relationship to the incidence of subtertian malaria. Transactions of the Royal Society of Tropical Medicine and Hygiene 48, 312–318 (1954)
——. Two lessons from the interface of genetics and medicine. Genetics 166, 1591–1599 (2004)
Centers for Disease Control and Prevention. Malaria. Centers for Disease Control and Prevention (2010)
Charache, S., et al. Effect of hydroxyurea on the frequency of painful crises in sickle-cell anemia. New England Journal of Medicine 332, 1317–1322 (1995)
Diggs, L. W., Ahmann, C. F., & Bibb, J. The incidence and significance of the sickle-cell trait. Annals of Internal Medicine 7, 769–778 (1933)
Hattori, Y., et al. Haplotypes of beta S chromosomes among patients with sickle-cell anemia from Georgia. Hemoglobin 10, 623–642 (1986)
Herrick, J. Peculiar elongated and sickle-shaped red blood corpuscles in a case of severe anemia. Archives of Internal Medicine 6, 517–521 (1910)
Ingram, V. M. Gene mutations in human haemoglobin: The chemical difference between normal and sickle-cell haemoglobin. Nature 180, 326–328 (1957)
——. Abnormal human haemoglobins. III. The chemical difference between normal and sickle-cell haemoglobins. Biochimica et Biophysica Acta 36, 402–411 (1959)
Kan, Y. W., & Dozy, A. M. Antenatal diagnosis of sickle-cell anaemia by DNA analysis of amniotic-fluid cells. Lancet 2, 910–912 (1978a)
——. Polymorphism of DNA sequence adjacent to human beta-globin structural gene: Relationship to sickle mutation. Proceedings of the National Academy of Sciences 75, 5631–5635 (1978b)
——. Evolution of the hemoglobin S and C genes in world populations. Science 209, 388–391 (1980)
Kulozik, A. E., et al. Geographical survey of beta S-globin gene haplotypes: Evidence for an independent Asian origin of the sickle-cell mutation. American Journal of Human Genetics 39, 239–244 (1986)
Labie, D., et al. Common haplotype dependency of high G gamma-globin gene expression and high Hb F levels in beta-thalassemia and sickle-cell anemia patients. Proceedings of the National Academy of Sciences 82, 2111–2114 (1985)
Lapoumeroulie, C., et al. A novel sickle-cell mutation of yet another origin in Africa: The Cameroon type. Human Genetics 89, 333–337 (1992)
Lettre, G., et al. DNA polymorphisms at the BCL11A, HBS1L-MYB, and beta-globin loci associate with fetal hemoglobin levels and pain crises in sickle-cell disease. Proceedings of the National Academy of Sciences 105, 11869–11874 (2008)
Lux, S. E., John, K. M., & Karnovsky, M. J. Irreversible deformation of the spectrin-actin lattice in irreversibly sickled cells. Journal of Clinical Investigation 58, 955–963 (1976)
Marotta, C. A., et al. Human beta-globin messenger RNA. III. Nucleotide sequences derived from complementary DNA. Journal of Biological Chemistry 252, 5040–5053 (1977)
Miller, B. A., et al. Molecular analysis of the high-hemoglobin-F phenotype in Saudi Arabian sickle-cell anemia. New England Journal of Medicine 316, 244–250 (1987)
Monteiro, C., et al. The frequency and origin of the sickle-cell mutation in the district of Coruche/Portugal. Human Genetics 82, 255–258 (1989)
Nagel, R. L., & Ranney, H. M. Genetic epidemiology of structural mutations of the beta-globin gene. Seminars in Hematology 27, 342–359 (1990)
Neel, J. V. The inheritance of sickle-cell anemia. Science 110, 64–66 (1949)
Oner, C., et al. Beta S haplotypes in various world populations. Human Genetics 89, 99–104 (1992)
Pagnier, J., et al. Evidence for the multicentric origin of the sickle-cell hemoglobin gene in Africa. Proceedings of the National Academy of Sciences 81, 1771–1773 (1984)
Pauling, L., et al. Sickle-cell anemia, a molecular disease. Science 110, 543–548 (1949)
Rahimi, Z., et al. Beta-globin gene cluster haplotypes in sickle-cell patients from southwest Iran. American Journal of Hematology 74, 156–160 (2003)
Rhinesmith, H. S., Schroeder, W. A., & Martin, N. The N-terminal sequence of the b chains of normal adult human hemoglobin. Journal of the American Chemical Society 80, 3358–3361 (1958)
Rhinesmith, H. S., Schroeder, W. A., & Pauling, L. A quantitative study of the hydrolysis of human dinitrophenyl (DNP) globin: The number and kind of polypeptide chains in normal adult human hemoglobin. Journal of the American Chemical Society 79, 4682–4686 (1957)
Rund, D., & Fucharoen, S. Genetic modifiers in hemoglobinopathies. Current Molecular Medicine 8, 600–608 (2008)
Schroeder, W. A., Munger, E. S., & Powars, D. R. Sickle-cell-anemia, genetic variations, and the slave-trade to the United States. Journal of African History 31, 163–180 (1990)
Sedgewick, A. E., et al. BCL11A is a major HbF quantitative trait locus in three different populations with beta-hemoglobinopathies. Blood Cells, Molecules, and Diseases 41, 255–258 (2008)
Steinberg, M. H. Predicting clinical severity in sickle-cell anaemia. British Journal of Haematology 129, 465–481 (2005)
——. SNPing away at sickle cell pathophysiology. Blood 111, 5420–5421 (2008)
Steinberg, M. H., et al. Effect of hydroxyurea on mortality and morbidity in adult sickle-cell anemia: Risks and benefits up to nine years of treatment. Journal of the American Medical Association 289, 1645–1651 (2003)
Uda, M., et al. Genome-wide association study shows BCL11A associated with persistent fetal hemoglobin and amelioration of the phenotype of beta-thalassemia. Proceedings of the National Academy of Sciences 105, 1620–1625 (2008)










