


 Figure 1
Figure 1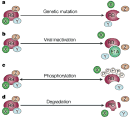















« Prev Next »
What drives cancer cells to grow and divide uncontrollably and to escape cell death? Studies of mutations in tumor suppressor genes have provided key answers to this question. Tumor suppressor genes often function to restrain inappropriate cell growth and division, as well as to stimulate cell death to keep our cells in proper balance. In addition, some of these genes are involved in DNA repair processes, which help prevent the accumulation of mutations in cancer-related genes. In this way, tumor suppressor genes act as "brakes" to stop cells in their tracks before they can take the road to cancer. Given this situation, loss of tumor suppressor gene function can be disastrous, and it often puts once-normal cells on the fast track to disease.
The Two-Hit Hypothesis: Knudson and RB1
Like all genes, tumor suppressor genes may undergo a variety of mutations; however, most loss-of-function mutations that occur in tumor suppressor genes are recessive in nature. Thus, in order for a particular cell to become cancerous, both of the cell's tumor suppressor genes must be mutated. This idea is known as the "two-hit" hypothesis, and it was first proposed by geneticist Alfred Knudson in 1971. Today, this hypothesis serves as the basis for researchers' understanding of how mutations in tumor suppressor genes drive cancer.
The two-hit hypothesis arose of out Knudson's interest in the genetic mechanisms underlying retinoblastoma, a childhood form of retinal cancer. Under normal circumstances, a population of cells in the developing eye, called retinoblasts, stops growing and dividing during embryogenesis and differentiates into retinal photoreceptor (light-capturing) cells and nerve cells. Typically, these differentiated cells do not divide very often, if ever. In the case of retinoblastoma, however, the retinoblasts fail to differentiate; thus, these cells continue to divide, forming tumors in the retina. If left untreated, the retinal tumor cells will eventually metastasize (spread) to other parts of the body.
Between 1944 and 1969, Knudson studied 48 patients with retinoblastoma who had been admitted to the hospital (Knudson, 1971). For each patient, Knudson tabulated the age at diagnosis, sex, family history, whether the tumor occurred in one eye (unilaterally) or in both eyes (bilaterally), and an estimation of the number of tumors present in each eye. He also supplemented this information with similar data from two previous publications (Vogel, 1957; Schappert-Kimmijser et al., 1966).
At the time of Knudson's study, researchers believed that retinoblastoma could be caused by either somatic or germ-line mutations. Knudson's combined data itself showed that retinoblastoma was caused by a germ-line mutation in approximately 40% of U.S. cases. However, Knudson was puzzled by the observation that some children with an affected parent were disease-free, but these unaffected individuals later bore children with retinoblastoma; this finding suggested that an individual could inherit a germ-line mutation but not have the disease. Knudson also noted that while the majority of children with an affected parent had bilateral tumors (25%–30%), some had only unilateral tumors (10%–15%). Furthermore, he determined that approximately 60% of retinoblastoma cases in the U.S. were unilateral and were not associated with a family history of the disease. These findings are summarized in Table 1; in the table, "hereditary" refers to those individuals with a family history of retinoblastoma.
| Bilateral | Unilateral | Total | |
| Hereditary | 25%–30% | 10%–15% | 35%–45% |
| Nonhereditary | 0 | 55%–65% | 55%–65% |
| Total | 25%–30% | 70%–75% | 100% |
| Table 1: Distribution of retinoblastoma cases by type and laterality | |||
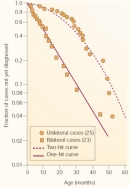
Next, Knudson used mathematics to determine whether the data followed a trend associated with a one-hit model or a two-hit model of gene mutation. In doing so, he used information from two groups; the first group consisted of 23 patients with bilateral hereditary retinoblastoma, and the second consisted of 25 patients with unilateral nonhereditary retinoblastoma. Because time is required for mutations to accumulate, Knudson examined the age at which the children in these two groups were diagnosed with retinoblastoma.

© 2001 Nature Publishing Group Knudson, A. Two genetic hits (more or less) to cancer. Nature Reviews Cancer 1, 160 (2001). All rights reserved. 
With this information in mind, Knudson took a closer look at his two groups of patients. Not surprisingly, he found that diagnosis for bilateral hereditary cases of retinoblastoma occurred at a younger age and showed a curve consistent with a single-mutation process. Furthermore, he noted that the rate of diagnosis for unilateral nonhereditary retinoblastoma was delayed relative to that of the bilateral cases and showed a curve consistent with a two-mutation process (Figure 1).
Based on calculations using this clinical data, Knudson concluded that retinoblastoma was caused by two mutations: one in each copy of a single tumor suppressor gene (now called RB1). He also estimated that each of the two mutations would occur at a rate of 2 x 10-7 per year. Patients who inherited an RB1 mutation would develop tumors earlier, and they would often develop more than one tumor. In contrast, individuals who did not inherit a mutation would almost always be affected by a single tumor. This statement, which Knudson called the two-mutation hypothesis (Figure 2), is now known as the two-hit hypothesis (Knudson, 1971).
Based on the predicted mutation rate, Knudson expected that many individuals in the general population would acquire a single somatic mutation in the RB1 gene over their lifetime, and that the retinas of most people would therefore likely contain small groups of retinoblasts that had received one "hit" in the RB1 gene. In order to become cancerous, each retinoblast with one mutant copy of the RB1 gene would need to acquire a mutation in the remaining wild-type copy of the gene. Most individuals who had one hit did not develop retinoblastoma, however, because most of their mutated cells had already differentiated and quit dividing before they could receive a second hit (Knudson, 2001).
Loss of Heterozygosity and Mechanisms of Tumor Suppressor Gene Inactivation
"Loss of heterozygosity" is a phrase often used to describe the process that leads to the inactivation of the second copy of a tumor suppressor gene. During this process, a heterozygous cell receives a second hit in its remaining functional copy of the tumor suppressor gene, thereby becoming homozygous for the mutated gene. Mutations that inactivate tumor suppressor genes, called loss-of-function mutations, are often point mutations or small deletions that disrupt the function of the protein that is encoded by the gene; chromosomal deletions or breaks that delete the tumor suppressor gene; or instances of somatic recombination during which the normal gene copy is replaced with a mutant copy.
RB1 and Other Tumor Suppressor Genes
Knudson developed the two-hit hypothesis long before the human genome was sequenced, and the RB1 gene was itself discovered in 1986. Researchers noticed that some cases of retinoblastoma were associated with a deletion of chromosome band 13q14 (Francke & Kung, 1976; Knudson et al., 1976) and then used restriction fragment length polymorphism (RFLP) analysis to isolate the RB1 gene (Friend et al., 1986).
With knowledge of the molecular identity of the retinoblastoma-associated gene in hand, researchers began to focus their efforts on the identification and characterization of RB1 function. Over the years, RB1 has been shown to block cell proliferation and cell division and to regulate cell death (Chau & Wang, 2003). RB1 collaborates with a number of cellular proteins in order to carry out its functions. Furthermore, RB1 function has been shown to be inactivated by four distinct mechanisms: genetic inactivation, sequestration of the RB1-encoded protein by viral oncoproteins, phosphorylation of the RB1-encoded protein, and degradation of the RB1-encoded protein (Figure 3). Many valuable insights into RB1 function have been gained through studies using mouse models in which the mouse Rb1 gene is deleted.
RB1 is one gene among a growing list of tumor suppressor genes. According to the American Cancer Society (2005), at least 30 different tumor suppressor genes have been identified, including those listed in Table 2. Many of these genes function to inhibit cell division and cell proliferation, stimulate cell death, and repair damaged DNA.
| Inherited Cancer | Mutated Tumor Suppressor Gene(s) | Gene Function(s) | Associated Noninherited Cancers |
| Retinoblastoma | RB1 | Cell division, DNA replication, cell death | Many different cancers |
| Li-Fraumeni syndrome (brain tumors, sarcomas, leukemia) | TP53 | Cell division, DNA repair, cell death | Many different cancers |
| Melanoma | CDKN2A (INK4A) | Cell division, cell death | Many different cancers |
| Colorectal cancer (due to familial polyposis) | APC | Cell division, DNA damage, cell migration, cell adhesion, cell death | Most colorectal cancers |
| Colorectal cancer (without polyposis) | MLH1, MSH2, MSH6 | DNA mismatch repair, cell cycle regulation | Colorectal, gastric, endometrial cancers |
| Breast and/or ovarian cancer | BRCA1, BRCA2 | Repair of double-stranded DNA breaks, cell division, cell death | Only rare ovarian cancers |
| Wilms' tumor | WT1, WT2 | Cell division, transcriptional regulation | Wilms' tumors |
| Nerve tumors (including brain) | NF1, NF2 | RAS-mediated signal transduction, cell differentiation, cell division, developmental processes | Small numbers of colon cancers, melanomas, neuroblastoma |
| Kidney cancer | VHL | Cell division, cell death, cell differentiation, response to cell stress | Certain types of kidney cancer |
|
Table 2: Commonly inherited cancers and associated tumor suppressor genes
(Adapted from American Cancer Society 2005) | |||
As evidenced by the growing number of identified tumor suppressor genes, our cells use many means to overcome genetic insults and precisely regulate their proliferation, growth, and death. Our increasing knowledge of tumor suppressor gene function will continue to enhance our ability to diagnose and more effectively treat cancers at the molecular level in the years to come.
References and Recommended Reading
American Cancer Society. Oncogenes and tumor suppressor genes, http://www.cancer.org/docroot/ETO/content/ETO_1_4x_oncogenes_and_tumor_suppressor_genes.asp (2005)
Chau, B. N., & Wang, J. Y. Coordinated regulation of life and death by RB. Nature Reviews Cancer 3, 130–138 (2003) doi:10.1038/nrc993 (link to article)
Francke, U., & Kung, F. Sporadic bilateral retinoblastoma and 13q- chromosomal deletion. Medical and Pediatric Oncology 2, 379–385 (1976)
Friend, S. H., et al. A human DNA segment with properties of the gene that predisposes to retinoblastoma and osteosarcoma. Nature 323, 643–646 (1986) (link to article)
Knudson, A. G. Mutation and cancer: Statistical study of retinoblastoma. Proceedings of the National Academy of Sciences 68, 820–823 (1971)
———. Two genetic hits (more or less) to cancer. Nature Reviews Cancer 1, 157–162 (2001) (link to article)
Knudson, A. G., et al. Chromosomal deletion and retinoblastoma. New England Journal of Medicine 295, 1120–1123 (1976)
Schappert-Kimmijser, J., et al. The heredity of retinoblastoma. Ophthalmologica 151, 197–213 (1966)
Vogel, F. New studies on the genetics of retinoblastoma: Glioma retinae. Zeitschrift für Menschliche Vererbungs und Konstitutionslehre 34, 205–236 (1957)










