


 Figure 3
Figure 3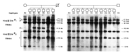















« Prev Next »
What if a simple blood test could tell you with absolute certainty that you would suffer from a deadly neurodegenerative disease late in life? What if the same test could tell you your chances of passing this disease on to your children? What if this disease had no possible treatment or cure? Such is the current state of Huntington's disease (HD), an adult-onset autosomal dominant disorder. Today, researchers can literally "measure" the HD-associated gene, called huntingtin (HTT), by determining the number of repeats of a set of three specific bases within this gene. An individual with 40 or more of these repeats in one copy of the HTT gene will certainly suffer from HD.
HD: Discovery, Inheritance Patterns, and Phenotypes
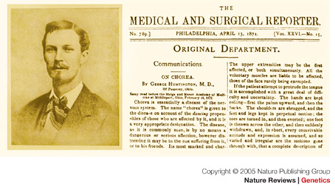
Figure 1: George Huntington as a young man.
The title page of George Huntington's 1872 paper in the Medical and Surgical Reporter.
© 2005 Nature Publishing Group Bates, G. P. The molecular genetics of Huntington disease — a history. Nature Reviews Genetics 6, 767 (2005). All rights reserved. 
HD is a rare, adult-onset, autosomal dominant, progressive neurodegenerative disease. George Huntington (Figure 1) was the first person to provide a comprehensive description of adult-onset HD in 1872; he was only 22 years old at the time. Huntington described the autosomal dominant inheritance pattern of this condition, which is accompanied by a loss of motor control leading to jerky movements, altered personality and psychiatric symptoms, and a decline in cognitive function.
HD symptoms typically manifest between 30 and 45 years of age, so the majority of HD patients have already had children before they are diagnosed, and they have thus passed the mutant HTT gene on to the next generation. Individuals carrying the mutated allele have a 50% chance of passing this allele to each of their children (Figure 2). In large families with a history of HD, the disease is likely to appear in every generation, because it is an autosomal dominant condition. Although HD phenotypes nearly always appear late in life, the dominant mutation in the HTT gene is present from birth.
Interestingly, J. Hoffman first reported a juvenile form of HD in 1888 through his studies of a three-generation family. Hoffman noted that the juvenile form of HD could be associated with characteristics that differed from the adult form of the disease, including rigidity, slow movements, and seizures. More than 100 years later, researchers realized that the juvenile form of HD is associated with a phenomenon called anticipation, in which HD phenotypes become more severe from one generation to the next. Anticipation occurs only when the mutant gene is inherited from the father. Currently, this inheritance pattern is not well understood.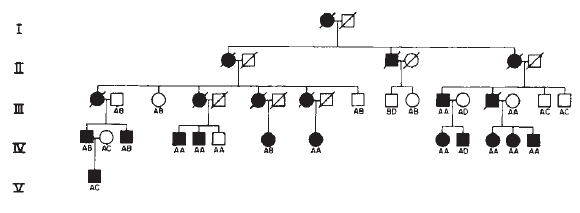
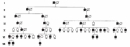
Figure 2: Pedigree of an American Huntington's disease family.
Symbols: circles, females; squares, males; a black symbol indicates that an individual is affected with Huntington's disease; a slashed symbol indicates that an individual is deceased.
© 1983 Nature Publishing Group Gusella, J. F. et al. A polymorphic DNA marker genetically linked to Huntington's disease. Nature 306, 235 (1983). All rights reserved.
The HTT Gene
Huntingtin (HTT) was the first disease-associated gene to be molecularly mapped to a human chromosome (Gusella et al., 1983). Ten years later, scientists identified the DNA sequence and determined the precise nature of the HD-associated mutation in HTT (MacDonald et al., 1993). It turns out that the HTT gene contains a region where the triplet nucleotide CAG is repeated several times. Moreover, the number of CAG repeats present in the HTT gene determines whether an individual will have HD. Individuals with six to 35 CAG repeats will be unaffected; individuals with 36-39 repeats will be at increased risk for HD; and individuals with 40 or more CAG repeats will definitely manifest disease phenotypes (MacDonald et al., 1993; Bates, 2005).
Today, scientists can use molecular genetic approaches to precisely determine the number of CAG repeats present in the HTT gene, and therefore accurately determine whether an individual will suffer from HD later in life. Unfortunately, a cure for HD does not currently exist. Thus, knowledge of the disease at the molecular genetic level cannot yet be met by any defensive measure at the phenotypic level.
Mapping Huntingtin (HTT)
As previously mentioned, HTT was first mapped to a specific chromosome in 1983. At that time, James F. Gusella and colleagues carried out a study to determine whether they could identify a DNA probe that would show an HD-associated restriction fragment length polymorphism (RFLP) when used in Southern blot analyses of chromosomal DNA digested with the restriction enzyme HindIII (palindromic recognition sequence 5'-AAGCTT-3'). The team identified one probe out of 12 tested, called G8, that showed a specific RFLP pattern associated with HD in two large families with a history of the disease (Gusella et al., 1983). Using the G8 probe, they next identified two HindIII sites (called H1 and H2) that were palindromic within this chromosomal region. DNA fragments at these sites vary in length among different HD lineages. Because researchers used two large pedigrees in these experiments, they were able to obtain statistical support for their discovery (Figure 3).
To obtain this pedigree information, Nancy Wexler, a prominent HD researcher, followed up on reports of a high incidence of HD in two Venezuelan communities located near Lake Maracaibo. Through her visits to the Lake Maracaibo region, Wexler found that the region's HD had originated with a single founder, suggesting that all affected individuals would carry the same original mutation as the founder. Over a period of more than 20 years, Wexler's research team was able to establish extensive pedigrees with medical histories. Together with a large HD-affected family in Ohio, the Venezuelan family provided the necessary variation among the H1 and H2 HindIII restriction site patterns that segregated with the HD in both families (Figure 4). This played a key role in mapping the HD gene (Gusella et al., 1983).
Using Human-Mouse Somatic Cell Hybrids
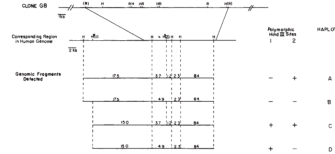
© 1983 Nature Publishing Group Gusella, J. F. et al. A polymorphic DNA marker genetically linked to Huntington's disease. Nature 306, 238 (1983). All rights reserved.
Although researchers had discovered that the G8 DNA probe was likely to correspond to a polymorphic chromosomal region located near the HD gene, they had no information about what human chromosome the G8 DNA probe came from. In order to map the G8 DNA probe to a specific human chromosome, researchers used a series of mouse cell lines, called human-mouse somatic cell hybrids, which were engineered to contain a known subset of human chromosomes.
By carrying out Southern blot experiments using the G8 DNA probe with chromosomal DNA from the human-mouse somatic cell hybrids, researchers could identify those cell lines that harbored human chromosomal DNA homologous to the G8 DNA probe. In addition, because they knew which human chromosomes were present in each of the human-mouse somatic cell hybrid lines, they could map the G8 DNA probe to a specific human chromosome. In these experiments, researchers found that the G8 DNA probe hybridized only to Southern blots using chromosomal DNA from human-mouse somatic cell hybrid lines that contained human chromosome 4. Therefore, researchers determined that the G8 DNA probe is located on human chromosome 4, and they concluded that the HD gene is located on chromosome 4 near the region corresponding to the G8 DNA probe (Figure 5; Gusella et al., 1983).
Identifying the HD Gene
After determining that the HD gene is located on chromosome 4, the members of the Huntington's Disease Collaborative Research Group (HDCRG) joined forces and spent the next 10 years working diligently to identify the HD gene and to determine the nature of the HD-associated mutation (MacDonald et al., 1993). The group eventually developed an extensive set of DNA probes that encompassed the region of chromosome 4 where the gene was believed to be located.
The members of HDCRG also predicted that HD was most likely associated with a mutation in a gene, rather than with a mutation in a noncoding region of chromosome 4. They focused on the genes in this region and identified IT15, which they showed was transcribed into mRNA. They then determined the DNA sequence of the IT15 gene and found that it was unlike any other previously identified human gene. Furthermore, they identified a region of the gene that contained a repeated DNA element consisting of three nucleotides, CAG, repeated 21 times near the beginning of the gene (MacDonald et al., 1993).
Discovering the HTT Mutation
In order to show that HD was indeed associated with the IT15 gene, the members of HDCRG needed to demonstrate that the IT15 gene was mutated in individuals with HD. The original IT15 DNA was derived from a healthy individual without HD. When researchers examined this same region of IT15 in other non-HD controls, they found that the number of CAG repeats varied from six to 35; they described this phenomenon as "instability of the trinucleotide repeat." Analysis of this same region in the IT15 gene in individuals with HD showed that these people always had 40 or more CAG repeats; in fact, the largest number of CAG repeats the researchers detected was 100 (MacDonald et al., 1993). The researchers thus concluded that the trinucleotide repeat expansion in the IT15 gene was responsible for HD, and IT15 is now called HTT (huntingtin).
The Future of Huntington’s Disease Management
Children of a parent with HD have a 50% chance of receiving the mutated HTT gene and thus developing the disease later in life. Today, through the use of PCR-based techniques, individuals at risk for HD can view their genetic blueprint and see their certain future. Whether they do so is a personal choice.
With the increasing number of HD-focused research foundations and our expanding knowledge of the molecular basis of HD, we can only hope that the collaborative efforts that led to the identification of the HTT gene and its mutations will continue into the future as we search for a cure for HD. Indeed, research teams throughout the world are currently exploring a wide range of molecular therapeutic approaches to combat this deadly disease.
References and Recommended Reading
Bates,
G. P. History of genetic disease: The molecular genetics of Huntington disease—A
history. Nature Reviews Genetics 6, 766–773 (2005) (link to article)
Gusella, J. F., et al. A polymorphic DNA marker genetically linked to Huntington's disease. Nature 306, 234–238 (1983) (link to article)
Hoffman, J. U'er Chorea chronica progressiva (Huntingtonsche Chorea, Chorea hereditaria). Virchows Archiv A 111, 513–548 (1888)
Huntington, G. On chorea. Medical and Surgery Reporter 26, 320–321 (1872)
MacDonald et al. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell 72, 971–983 (1993)










