

















« Prev Next »
Even though both parents contribute equally to the genetic content of their offspring, a developmental process called genomic imprinting sometimes leads to the exclusive expression of specific genes from only one parent. This process was first described in 1984, when two laboratories discovered a mark, or "imprint," that differentiates between certain genes on the maternal and paternal chromosomes and results in the expression of only one copy of those genes in the offspring. The genes in imprinted areas of an organism's genome are expressed depending on the parent of origin. As a result, the inheritance of both the maternal and paternal genes is required for normal development to proceed (McGrath & Solter, 1984; Surani et al., 1984).
To understand how imprinting works, rather than looking at the entire genome, consider the effect of this process on smaller chromosomal regions and single genes. For many diploid genes, even if the copy you inherited from one parent is defective, you have a substitute allele from your other parent. However, in the case of imprinting, even though there are two copies of the gene, it is as if you are haploid for this gene because only one copy is expressed. In other words, there is no substitute allele, which makes imprinted genes more vulnerable to the negative effects of mutations. Additionally, genes and mutations that might normally be recessive can be expressed if a gene is imprinted and the dominant allele is silenced (Jirtle & Weidman, 2007).
Diseases Related to Imprinting
As you might expect, it is therefore possible for diseases to occur due to deletions or mutations in imprinted genes. Diseases can also result from uniparental disomy, or the inheritance of two copies of a chromosome from one parent and no copy from the other parent, when the involved gene is imprinted. Additionally, diseases are possible when there are mutations in genes responsible for the imprinting process and when the imprint is not set correctly. Some examples of genetic diseases related to errors in the imprinting of specific genes and chromosomal regions include Prader-Willi syndrome, Angelman syndrome, and several types of cancer.
Prader-Willi Syndrome and Angelman Syndrome
Prader-Willi syndrome was first described by John Langdon Down (who also identified Down syndrome) in 1887, and later reported by Andrea Prader, Alexis Labhart, and Heinrich Willi in 1956. This disorder occurs in approximately one in 20,000 births and is associated with behavioral and cognitive problems, including mental retardation, deficiencies in sexual development and growth, hyperphagia, and obesity (Prader et al., 1956; Falls et al., 1999).
In 1965, Dr. Harry Angelman was the first to report the symptoms of Angelman syndrome. The disorder occurs in approximately one in 15,000 births, and the syndrome is characterized by developmental deficiencies, mental retardation, sleep disorders, seizures, ataxia, hyperactivity, and a happy disposition with outbursts of laughter (Angelman, 1965; Falls et al., 1999).
Prader-Willi syndrome and Angelman syndrome were the first imprinting diseases discovered in humans. The symptoms of these two disorders are very different, but scientists discovered that both conditions are caused by indistinguishable deletions in chromosome 15 in the 15q11-q13 region (Knoll et al., 1989). What distinguishes these disorders is the parental origin of the affected chromosome. Specifically, Prader-Willi syndrome is caused by the loss of a group of paternally inherited genes on chromosome 15 (Butler et al., 1986; Nicholls et al., 1989a, 1989b). In contrast, Angelman syndrome is caused by the loss of a maternally inherited gene in the same region of chromosome 15. Researcher Joan Knoll and her colleagues thus concluded that both Prader-Willi syndrome and Angelman syndrome were due to defects in imprinted genes, as the imprinted region of chromosome 15 normally contains genes that are either paternally or maternally expressed (Knoll et al., 1989).
In most cases (60%-70%), Prader-Willi syndrome is caused by deletions of a genetic region that includes the small nuclear ribonucleoprotein polypeptide N gene, the necdin gene, and possibly other genes (Robertson, 2005). In the remaining 20%-30% of Prader-Willi patients, the disorder occurs because the affected individual has two copies of maternal chromosome 15 and no copy of the corresponding paternal chromosome (Robertson, 2005). This condition is called maternal unipaternal disomy. It is not yet known how the loss of expression of these paternally imprinted genes mechanistically results in Prader-Willi syndrome.
The genetic errors associated with Angelman syndrome are, of course, somewhat different. In particular, Tatsuya Kishino et al. (1997) showed that Angelman syndrome is due to the loss of expression of a single maternally expressed gene in the region, called UBE3A. The UBE3A gene encodes a protein called E3 ubiquitin ligase, which is involved in targeting proteins for degradation, and it is only imprinted in the brain. The loss of UBE3A may result in abnormalities in normal protein degradation during brain development, thereby causing Angelman syndrome (Kishino et al., 1997). In fact, in most cases (65%-70%), Angelman syndrome is caused by maternally derived deletions of the UBE3A gene (Robertson, 2005). However, this condition can also be caused by paternal unipaternal disomy (wherein embryos inherit both copies of chromosome 15 from their father), mutations in the UBE3A gene, and imprinting defects, such as the loss of maternal DNA methylation (Robertson, 2005).
Cancer
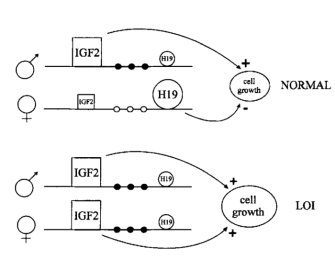
Figure 1: Model of loss of imprinting of IGF2, H19 and methylation of the H19 promoter in Wilms' tumor.
In normal cells, the paternal IGF2 and maternal H19 genes are expressed (shown large). Several sites upstream of H19 are methylated on the paternal allele (filled circles) and unmethylated on the maternal allele (open circles). In tumors with LOI, the maternal chromosome reverses to a paternal epigenotype, with a paternal pattern of methylation of the H19 promoter, IGF2 turned on, and H19 turned off, causing increased cell growth. LOI of H19 on the maternal chromosome, when it occurs, could occur independently or could be influenced by events in the paternal chromosome.
© 1994 Nature Publishing Group Steenman, M. et al. Loss of imprinting of IGF2 is linked to reduced expression and abnormal methylation of H19 in Wilms' tumor. Nature Genetics 7, 437 (1994). All rights reserved. 
In addition to these syndromes, imprinting has been linked to certain cancers. For instance, Wilms' tumor is a type of embryonic kidney cancer that is associated with the IGF2/H19 locus on chromosome 11. H19 is a noncoding RNA of unknown function with properties that can suppress growth. IGF2 codes for insulin-like growth factor 2, a growth factor highly expressed in many types of tumors. These two genes are both imprinted, and normally, only the maternal copy of H19 and the paternal copy of IGF2 are expressed (Steenman et al., 1994). In cancer cells, however, both H19 and IGF2 typically lose their imprinting (Moulton et al., 1994; Steenman et al., 1994). Scientists hypothesize that if the function of H19 is to turn off IGF2 expression, then the loss of H19 expression could result in overexpression of IGF2, thus leading to tumorigenesis (Steenman et al., 1994).
Research has helped support this conclusion, as Wilms' tumor cells have shown loss of imprinting of the maternal chromosome and a switch to the paternal pattern of methylation (Steenman et al., 1994). Moreover, Thomas Moulton and colleagues showed that H19 mRNA expression was decreased at least twentyfold in most Wilms' tumors, indicating that H19 is inactivated in tumor cells (Moulton et al., 1994). This results in the overexpression of IGF2 and reduced expression of H19. Because H19 functions to slow cell growth and IGF2 stimulates cell growth, loss of imprinting at the H19/IGF2 locus results in uncontrolled cell growth that can lead to tumor formation (Figure 1; Steenman et al., 1994).
Loss of imprinting (i.e., the loss of normal allele-specific gene expression) can also result in cancer when an imprinted, normally silent allele that provides cells with a growth advantage is activated, resulting in uncontrolled cell growth and division. For example, along with Wilms' tumor, loss of imprinting of the IGF2 gene is associated with many other types of cancer, including lung, colon, and ovarian tumors (Robertson, 2005). Cancers can also form in cases in which a tumor suppressor gene is imprinted, and the single expressed copy of the tumor suppressor is mutated or loses its function (Jirtle & Weidman, 2007).
Imprinting Disorders are On the Rise
In recent years, there has been an increase in the incidence of various imprinting disorders, especially among children conceived using assisted reproductive technologies, such as in vitro fertilization (Maher et al., 2003; Cox et al., 2002). Because imprinting occurs during gametogenesis, there are growing concerns that various elements of reproductive assistance procedures prevent genes from being imprinted properly or stably transferred during early embryonic development (Clayton-Smith, 2003; Jirtle & Weidman, 2007). Understanding the process of imprinting and identifying conditions that interfere with normal imprinting may thus help reduce the incidence of these conditions.
References and Recommended Reading
Angelman, H. "Puppet children": A report of three cases. Developmental Medicine and Child Neurology 7, 681–688 (1965)
Butler, M. G., et al. Clinical and cytogenetic survey of 39 individuals with Prader-Labhart-Willi syndrome. American Journal of Medical Genetics 23, 793–809 (1986)
Clayton-Smith, J. Genomic imprinting as a cause of disease. British Medical Journal 327, 1121–1122 (2003) doi:10.1136/bmj.327.7424.1121
Cox, G. F., et al. Intracytoplasmic sperm injection may increase the risk of imprinting defects. American Journal of Human Genetics 71, 162–164 (2002)
Down, J. L. Mental Affections of Childhood and Youth (London, Churchill, 1887)
Falls, J. G., et al. Genomic imprinting: Implications for human disease. American Journal of Pathology 154, 635–647 (1999)
Jirtle, R. L., & Weidman, J. R. Imprinted and more equal. American Scientist 95, 143–149 (2007)
Kishino, T., et al. UBE3A/E6-AP mutations cause Angelman syndrome. Nature Genetics 15, 70–73
(1997) doi:10.1038/ng0197-70 (link to article)
Knoll, J. H., et al. Angelman and Prader-Willi syndromes share a common chromosome 15 deletion but differ in parental origin of the deletion. American Journal of Medical Genetics 32, 285–290 (1989)
Maher, E. R., et al. Beckwith-Wiedemann syndrome and assisted reproduction technology (ART). Journal of Medical Genetics 40, 62–64 (2003)
McGrath, J., & Solter, D. Completion of mouse embryogenesis requires both the maternal and paternal genomes. Cell 37, 179–183 (1984)
Moulton, T., et al. Epigenetic lesions at the H19 locus in Wilms' tumor patients. Nature Genetics 7, 440–447 (1994) (link to article)
Nicholls, R. D., et al. Genetic imprinting suggested by maternal heterodisomy in nondeletion Prader-Willi syndrome. Nature 342, 281–285 (1989a) (link to article)
———. Restriction fragment length polymorphisms within proximal 15q and their use in molecular cytogenetics and the Prader-Willi syndrome. American Journal of Medical Genetics 33, 66–77 (1989b)
Prader, A., et al. Ein Syndrom von Adipositas, Kleinwuchs, Kryptorchismus und Oligophrenie nach Myatonieartigem Zustand im Neugeborenenalter. Schweizerische Medizinische Wochenschrift 86, 1260–1261 (1956)
Robertson, K. D. DNA methylation and human disease. Nature Reviews Genetics 6, 597–610 (2005) doi:10.1038/nrg1655 (link to article)
Steenman, M. J., et al. Loss of imprinting of IGF2 is linked to reduced expression and abnormal methylation of H19 in Wilms' tumor. Nature Genetics 7, 433–439 (1994) (link to article)
Surani, M. A. H., et al. Development of reconstituted mouse eggs suggests imprinting of the genome during gametogenesis. Nature 308, 548–550 (1984) doi:10.1038/308548a0 (link to article)










