



 Table 1
Table 1
















« Prev Next »
Human somatic cells contain 23 pairs of chromosomes, and we inherit one set of 23 chromosomes from each of our parents. Chromosomes are built using four different types of DNA bases: adenine (A), cytosine (C), guanine (G), and thymine (T). The human genome is defined by more than 3 billion base pairs of DNA that make up our 46 chromosomes; these DNA base pairs are organized in a specific order and serve as the blueprint that distinguishes humans from other species. Studies of the human genome have shown that between 20,000 and 25,000 genes reside along our chromosomes. Although redundancy is built into the human genome, our gene repertoire is exquisitely regulated in order to maintain cell function by copying to RNA, which in turn determines the formation of proteins. Thus, even a single base-pair mutation in one of our genes can lead to an altered protein and consequently disease.
Although the complexities of human biology suggest an equally complex genome, the human genome is actually quite similar to that of the mouse. The mouse genome is built of 2.7 billion base pairs of DNA, and current estimates suggest that the mouse genome contains at least 28,972 genes, of which at least 16,927 have homologues (called orthologues) in humans. In fact, 846 human genetic diseases have been reproduced in mouse strains (called mouse genotypic models) that carry a mutation in the related mouse gene.
So, what distinguishes humans from other organisms? What makes us human is a combination of the following:
- The ability of a single gene product (protein) to perform multiple functions due to overlapping roles in different pathways
- The ability of our genes to undergo different types of RNA splicing to produce gene variants with alternative functions
- The important functions carried out by chromosomal DNA segments located between genes
- Epigenetic regulation
Roll Call: From Genes to Disease
Because we inherit one set of 23 chromosomes from each of our parents, our somatic cells contain two copies of each of these 20,000 to 25,000 genes. Most of the human genome is nearly identical from one person to another. In fact, on average, a randomly chosen 1,200-base-pair segment of human chromosomal DNA contains only one base pair that varies between two unrelated individuals. The vast majority of these DNA variants are not deleterious, but some specific alterations in the human DNA sequence are associated with disease.
Indeed, recent estimates suggest that mutations in at least 383 human genes lead to known phenotypes, and that more than 2,336 human phenotypes are understood at the molecular level. However, the molecular basis of 1,630 confirmed Mendelian phenotypes is not known, and an additional 2,081 phenotypes are suspected to exhibit Mendelian inheritance (Online Mendelian Inheritance in Man, 2008). Although genetic "pills" do not yet exist to target the vast majority of disease-related mutations, our knowledge of the human genome sequence has opened new doors to the development of gene-based therapeutic approaches (Brinkman et al., 2006; O'Connor & Crystal, 2006).
Treating Disease Before Knowing the Genes Involved
Long before the human genome was sequenced, doctors were already treating many hereditary forms of human disease with surprising success. Many of these diseases were metabolic disorders, and doctors were able to determine whether a metabolic product was accumulating to harmful levels, or whether a key intermediary in a metabolic pathway was missing, even though the disease-associated gene was not yet known. In some cases, such disease phenotypes can be kept in check by dietary modification or by providing a missing protein. In other cases, surgical approaches can be used to repair or replace an organ or tissue damaged by disease.
Metabolic Manipulation
Physicians have developed approaches to regulate the metabolic pathways associated with a number of disorders, including phenylketonuria (PKU), sickle-cell anemia, hereditary angioedema, familial hypercholesterolemia, thalassemia, and many others. Often, this form of "metabolic manipulation" can be accomplished by modifying a patient's diet. For example, patients with PKU accumulate high levels of a protein building block, called phenylalanine, in their bloodstream. Doctors are able to diagnose PKU using a simple heel-prick blood test to detect high levels of phenylalanine by three days of age. Once diagnosed, infants with PKU are fed a diet low in protein and phenylalanine, which helps prevent PKU-associated cognitive decline.
In other cases, metabolic manipulation involves the use of small molecules or drugs to target the activity of proteins linked to disease. For instance, familial hypercholesterolemia is associated with high levels of "bad" (LDL) cholesterol and early heart disease; in this case, treatment includes both dietary modifications (a diet low in cholesterol) and the administration of a class of drugs called statins. These medications inhibit the activity of an enzyme (HMG CoA reductase) involved in a rate-limiting step of cholesterol biosynthesis.
Hemoglobin is the iron-containing protein that carries oxygen in our red blood cells. Humans express different forms of hemoglobin during development. Hemoglobin is built of four protein subunits: two ά-subunits and a second pair of subunits, which vary by age. The main adult form of hemoglobin consists of two ά-subunits and two β-subunits (ά2β2), whereas the fetal form of hemoglobin consists of two ά-subunits and two γ-subunits (ά2γ2). Sickle-cell anemia is due to a mutation in the gene encoding the β-subunit of hemoglobin (called HBB), which leads to an abnormal structure of the β-subunit protein chain and the sickle-cell phenotype (Weatherall, 2003). In the 1980s, researchers discovered that treatment with a drug called hydroxyurea, which increases levels of the γ-subunit of fetal hemoglobin, leads to a 50% reduction in the painful sickle-cell crises associated with sickle-cell anemia (Platt et al., 1984). Hydroxyurea is still used to treat sickle-cell anemia today.
Protein Augmentation
In another approach called protein augmentation, physicians treat patients by providing them with a purified form of the missing, defective, or depleted protein. This protein-add-back approach has been used to successfully treat patients suffering from a wide range of diseases, including various membrane transport disorders (cystic fibrosis), coagulation disorders (hemophilia A, hemophilia B, and Von Willebrand disease), emphysema (ά1-antitrypsin deficiency), immune deficiency (severe combined immune deficiency), endocrine disorders (growth hormone deficiency, congenital leptin deficiency, and congenital neurogenic diabetes insipidus), and lysosomal storage disorders (Gaucher's disease type I, Fabry disease, mucopolysaccharidosis I, mucopolysaccharidosis II, mucopolysaccharidosis VI, and Pompe's disease).
For example, the gastrointestinal symptoms that accompany cystic fibrosis can be corrected by protein augmentation through the administration of pancreatic enzymes. Similarly, individuals with growth hormone deficiency can be treated with purified growth hormone to restore normal growth. In addition, symptoms associated with Pompe's disease, including reduced heart, lung, and skeletal function, can be improved by treatment with acid ά-glucosidase enzyme.
Protein augmentation requires that the protein be added to the outside of cells (i.e., the extracellular space). Therefore, this approach works best for replacing proteins that are normally present in the extracellular space. Protein augmentation approaches are often less effective if the missing protein is normally located inside of a cell, or if it is normally targeted to a specific intracellular organelle. Indeed, it can be difficult to ensure that a protein added to the outside of a cell will be taken up by the cell and targeted correctly to its normal location or organelle.
In some cases, the success of protein augmentation depends on how well the protein is delivered to the organ(s) in which its function is required. The brain is a particularly difficult organ to target, because the access of proteins is limited by a membrane structure called the blood-brain barrier (BBB), which inhibits the passage of proteins and other chemicals from the bloodstream into the brain. The eye also presents challenges to drug access from the bloodstream. However, therapeutic agents can be delivered directly to the eye; this is not possible with the brain.
Surgical Approaches
Although more invasive, organ transplantation is also used to treat certain genetic diseases that affect particular organs. Unless the organ donor and the organ recipient are monozygotic twins, the chromosomal DNA sequence of the donor will be different from that of the recipient. Despite these differences, organ transplantation remains a viable therapy that continues to be used widely to this day.
In still other cases, surgery can be used to repair an organ or tissue that has been targeted by disease. For example, cleft lip, with or without cleft palate, is the fourth most common birth defect in the U.S., and it affects 1 in every 700 babies born each year. Cleft lip and/or cleft palate arise early during pregnancy while a baby is developing in the uterus; the malformations occur when there is not enough tissue in the lip or mouth area to permit joining of the tissue, or when the fusion of the lateral structures of the face does not occur properly. Cleft lip occurs when the two sides of the upper lip are separated, and cleft palate occurs when either the hard palate or the soft palate is separated. Surgical approaches can be used to effectively repair both cleft lip and cleft palate defects.
Putting the Human Genome to Work: Using Genes to Treat Disease
The identification of disease-causing mutations can be a rich source of opportunity for identifying new disease treatments. After identifying a disease-associated mutation, scientists can study how the function of the corresponding gene product (protein) is altered. With this information in hand, three main gene-based therapeutic approaches are currently being pursued:
- Gene-transfer approaches, in which a wild-type copy of the mutated gene is delivered
- RNA modification therapy, in which the mRNA encoded by a mutant gene is targeted
- Stem cell therapy, in which human stem cells are used to repair disease-damaged tissue
Gene Transfer
Gene-transfer approaches are based upon the simple concept that if a disease is due to a recessive loss-of-function mutation in a single gene, then adding back the wild-type gene should restore normal function and alleviate the disease phenotype. So far, two strategies have been used to deliver wild-type genes (O'Connor & Crystal, 2006):
- Ex vivo approaches, which require the use of a cell population that can be removed from a patient, genetically altered to express the wild-type gene, and then delivered back into the patient
- In vivo approaches, which deliver the wild-type gene directly into a patient's affected cell population, tissue, or organ
Although the concept is simple, gene-transfer approaches have presented many clinical challenges. For example, both ex vivo and in vivo approaches require two key components: a gene expression cassette, consisting of a wild-type gene and some extra DNA sequences that permit the gene's expression in human cells, and a delivery system to introduce the wild-type gene into cells. The gene delivery system can be a cell membrane–like lipid carrier, or it can be derived from a virus. Table 1 shows some examples of gene-transfer clinical trials and the gene delivery systems employed, including delivery of the factor VIII (F8) gene to fibroblasts for the treatment of hemophilia A, delivery of the CFTR gene to the nasal and airway epithelium for the treatment of cystic fibrosis, delivery of the ornithine transcarbamylase (OTC) gene to the liver for the treatment of ornithine transcarbamylase deficiency, delivery of the glucocerebrosidase (GBA) gene to blood and bone marrow cells for the treatment of Gaucher's disease, and delivery of the ά1-antitrypsin (SERPINA1) gene for the treatment of ά1-antitrypsin deficiency. Additional information about clinical trials using gene delivery approaches to treat human disease can be found on the Gene Therapy Clinical Trials Worldwide website.
Figure 1a shows a typical gene expression cassette. The wild-type gene (called a transgene) has a promoter sequence attached to its 5′ end, which allows it to be recognized by the transcription machinery of the cell, and a polyadenylation signal at its 3′ end, which leads to the production of a poly-A tail and improved stability of the resulting mRNA. Current approaches typically use the cDNA corresponding to the wild-type gene, which does not contain introns. Figures 1b through 1f show some of the different methods used for gene delivery. Each gene delivery system has its own preferred type of expression cassette and its own maximal capacity in terms of the size of the transgene it can carry, as shown.
In order to successfully deliver the gene of interest, both lipid carriers and viruses must facilitate cell entry by breaking through the target cell's plasma membrane, and they must allow the expression cassette to enter the cell nucleus so that it can be transcribed into mRNA. Once in the nucleus, the expression cassette can either undergo random insertion into the cell's genome, or it can be maintained extrachromosomally within the nucleus, depending upon the delivery system. If chromosomally integrated, the transgene will be expressed continuously; however, the random nature of chromosome integration can lead to problems, such as cancer, if the gene is inserted in such a way that it disrupts the function of another gene. On the other hand, if the expression vector is maintained extrachromosomally, expression of the transgene can be lost over time due to cell division.
Researchers must also strike a balance in terms of the expression levels of the transgene. Specifically, expression must be sustained at high enough levels to rescue disease-related phenotypes, but it must also be maintained at low enough levels to prevent an immune response by the patient.
Targeting RNA to Treat Dominant Genetic Diseases
Gene-transfer approaches are particularly useful when a disease-associated mutation encodes a protein with decreased function, called a loss-of-function mutation; in this case, normal cellular function is restored when a wild-type copy of the gene is introduced, because loss-of-function mutations are usually recessive. However, human disease can also be associated with dominant mutations in genes that encode hyperactive proteins, called gain-of-function mutations. Furthermore, human disease can be associated with dominant mutations in which the mutant proteins interfere with the function of wild-type proteins, called dominant negative mutations.
In the case of a dominant mutation, introduction of the corresponding wild-type gene is usually not sufficient to rescue the disease-associated phenotype(s); rather, researchers would prefer to "turn off" expression of the mutant gene, or to inhibit the function of the mutant protein it encodes. To achieve this goal, researchers have turned their attention to RNA-based approaches, which target the RNA (either pre-mRNA or mRNA) transcribed from the dominant negative gene and effectively inhibit expression of the mutant protein.
Figure 2 shows examples of RNA-based strategies for the treatment of disease. Five approaches have been used experimentally to modify RNA levels: antisense oligonucleotides (ASO), RNA interference (RNAi), trans-splicing, segmental trans-splicing, and ribozymes.
ASO strategies use short single-stranded DNA (ssDNA) molecules, usually between 18 and 30 bases long, which are complementary to the mRNA to be targeted (Figure 2a). The ssDNA binds to the target mRNA, and the resulting DNA-RNA hybrid molecule is then degraded by the intracellular enzyme ribonuclease H (RNase H).
RNAi involves the use of double-stranded RNA molecules (dsRNA), typically 22 base pairs long, corresponding to a region of the target gene (Figure 2b). The dsRNA is processed within the cell in such a way that it becomes part of an RNA-induced silencing complex (RISC) that recognizes and degrades the corresponding target mRNA.
Trans-splicing is a gene-transfer approach that targets a pre-mRNA containing a disease-associated mutation within one of its exons (Figure 2c). In this case, the transgene is used to replace the exon carrying the disease-associated mutation (exon C* in Figure 2c) with a wild-type copy of the exon. The transgene contains a hybridization domain, which is complementary to a region of the 5′ flanking intron between the donor and branch-point sites for RNA splicing, followed by the splicing branch point, the splice acceptor site, the wild-type exon sequence, and the rest of the gene. Trans-splicing leads to the production of a wild-type copy of the mature mRNA and thus a corresponding wild-type protein.
Segmental trans-splicing is an approach used to get around the size limitations associated with gene-transfer methods that involve vectors. (Sometimes, a given cDNA is too large to be carried within a single viral vector.) In this case, the gene is divided into two smaller pieces, which are delivered together using two separate gene-transfer vectors (Figure 2d). The vector carrying the second half of the gene includes a hybridization domain complementary to an intron located at the 3′ end of the first half of the gene, similar to that described for trans-splicing. In this case, trans-splicing leads to the production of a mature mRNA encoding the full length of the wild-type protein of interest.
Ribozymes are RNA molecules with inherent catalytic activity that recognize a particular mRNA and cleave it (Figure 2e). Ribozymes containing a hybridization domain followed by a ribozyme nucleolytic motif that recognizes a target mRNA and the corresponding wild-type gene sequence can be used to selectively cleave a target mRNA that contains a mutation after the ribozyme cleavage site. Once the target gene is cleaved, the ribozyme-derived hybridization motif binds, and RNA splicing leads to the formation of a wild-type copy of the mature mRNA.
Many of these RNA-based strategies have been developed in recent years, and numerous questions remain regarding the cellular mechanisms involved in mRNA targeting. Furthermore, researchers must exercise caution with respect to the specificity of any given mRNA-targeting approach, whether ASO, RNAi, trans-splicing, or ribozyme based, to ensure that only the mRNA of interest is targeted.
Stem Cell Therapy
On the genetic horizon, the modern-day equivalent of organ transplantation is likely to be the use of stem cell therapy. Unlike organs, which are built of specialized mature cells with tissue-specific characteristics and a very limited ability to divide, stem cells are immature cells that have not yet specialized and that have the capacity to divide and mature into a wide variety of tissue types.
Stem cells naturally occur in two forms: embryonic stem cells and somatic stem cells. Embryonic stem cells are derived from a specific group of cells within an embryo. They are capable of unrestricted cell divisions (i.e., they are immortal) and are pluripotent, which means that they are able to become nearly any cell type imaginable as long as they are provided with the appropriate environment. Ethical concerns regarding the use of human embryos as a source of stem cells, as well as technical difficulties in obtaining and culturing these cells, have hampered the clinical use of embryonic stem cells.
Somatic stem cells are derived from a specific group of cells within an adult tissue that serve to renew the tissue cell populations over time. Unlike pluripotent cells, somatic stem cells are more restricted in terms of the type of cells they can become after they divide; their fate depends upon the tissue type from which they are derived. Figure 3 shows the differences between embryonic stem cells and somatic stem cells in terms of how they are derived and their ability to differentiate.
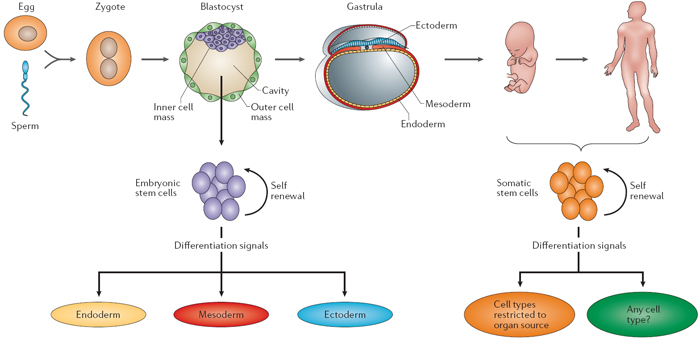
Figure 3: Differences between embryonic and somatic stem cells.
Differences between embryonic and somatic stem cells.
© 2008 Nature Education All rights reserved. 
One of the most exciting breakthroughs in stem cell research occurred recently and has been reproduced in labs across the world: the ability to convert somatic cells into pluripotent stem cells (Takahashi et al., 2007; Yu et al., 2007; Lowry et al., 2008; Park et al., 2008b). Scientists have identified a set of four genes (OCT4/POU5F1, SOX2, KLF4, and c-MYC/MYC) that encode transcription factors that, when expressed at the same time, can convert skin cells (dermal fibroblasts) taken from an adult human into induced pluripotent stem cells (iPS cells) that are phenotypically indistinguishable from human embryonic stem cells in terms of their gene expression, cell surface markers, and cellular morphology. Like human embryonic stem cells, the iPS cells are immortal, are pluripotent, and express genes characteristic of all three embryonic germ cell layers (endoderm, ectoderm, and mesoderm) when induced to differentiate. Both opponents and proponents of human stem cell research have warmly welcomed the promise of somatic cell–derived iPS cell lines.
The ability to produce iPS cell lines from somatic cells was heralded as an invaluable tool for understanding the underlying mechanisms associated with disease and for developing novel approaches to the treatment of human disease. Recently, a team of researchers at Harvard University applied this technique and established a panel of human disease–specific iPS cell lines (Park et al., 2008a); in this case, the investigators expressed three (OCT4/POU5F1, SOX2, and KLF4), four (OCT4/POU5F1, SOX2, KLF4, and c-MYC/MYC), or five (OCT4/POU5F1, SOX2, KLF4, c-MYC/MYC and NANOG) transcription factor genes.
To generate disease-specific iPS cell lines, researchers collected skin cells and bone marrow–derived mesenchymal cells from patients with one of ten different diseases, including adenosine deaminase deficiency-related severe combined immunodeficiency (ADA-SCID), Shwachman-Bodian-Diamond syndrome (SBDS), Gaucher's disease (GD) type III, Duchenne muscular dystrophy (DMD), Becker muscular dystrophy (BMD), Parkinson's disease (PD), Huntington's disease (HD), juvenile-onset type 1 diabetes mellitus (JDM), Down syndrome/trisomy 21 (DS), and Lesch-Nyhan syndrome (carrier). Similar to the original iPS cell lines, the disease-specific iPS cell lines were immortal, pluripotent, and capable of expressing genes corresponding to all three embryonic cell layers when induced to differentiate (Park et al., 2008a).
Table 2 summarizes the panel of disease-specific iPS cell lines and provides details about the corresponding mutated somatic cell types from which they were derived, as well as the age and sex of the somatic cell donor (Park et al., 2008a). These cell lines are freely accessible to researchers worldwide, and they will serve as a strong foundation for future studies aimed at the eradication of these devastating diseases. Due to the viral-based methods used to deliver the transcription factor genes into the somatic cells, the resulting iPS cell lines cannot currently be used to treat human patients. Nevertheless, these cell lines are an invaluable resource for future investigation. With these disease-specific iPS cell lines in hand, researchers will be able to carry out experiments to better understand disease pathology and to develop effective gene-transfer techniques, RNA-based therapies, and drug-screening approaches to target disease phenotypes.
Table 2: iPS Cells Derived from Somatic Cells of Patients with Genetic Disease
| Name | Disease | Molecular Defect | Donor Cell | Age | Sex |
| ADA | ADA-SCID | GGG > AGG, exon 7 and Del(GAAGA) exon 10, ADA gene | Fibroblast | 3M | Male |
| GD | Gaucher's disease type III | AAC > AGC, exon 9, G-insertion, nucleotide 84 of cDNA, GBA gene | Fibroblast | 20Y | Male |
| DMD | Duchenne muscular dystrophy | Deletion of exons 45–52, dystrophin gene | Fibroblast | 6Y | Male |
| BMD | Becker muscular dystrophy | Unidentified mutation in dystrophin gene | Fibroblast | 38Y | Male |
| DS1 | Down syndrome | Trisomy 21 | Fibroblast | 1Y,1M | Male |
| PD | Parkinson's disease | Multifactorial | Fibroblast | 57Y | Male |
| JDM | Juvenile diabetes mellitus | Multifactorial | Fibroblast | 42Y | Female |
| SBDS | Shwachman-Bodian-Diamond syndrome | IV2 + 2T > C and IV3 - 1G > A, SBDS gene | Bone marrow mesenchymal cells | 4M | Male |
| HD | Huntington's disease | 72 CAG repeats, huntingtin gene | Fibroblast | 20Y | Female |
| LNSc | Lesch-Nyhan syndrome (carrier) | Heterozygosity of HPRT1 | Fibroblast | 34Y | Female |
Reproduced from Park et al., 2008a
Looking Ahead: Gene-Inspired Drug Design and Multimodal Therapies
Researchers' ever-increasing knowledge of human genes and their disease-associated mutations has inspired new approaches to drug design and discovery. By understanding the underlying molecular mechanisms linked to disease, investigators can better target the activities of the enzymes, cell surface receptors, secreted proteins, intracellular signaling proteins, and transcription factors that regulate disease-associated phenotypes. As gene-based therapeutics continue to evolve, multimodal approaches to human disease will emerge. The future of genetic medicine will require collaboration and multidisciplinary approaches, which will most certainly be accompanied by unexpected, life-changing discoveries.
References and Recommended Reading
Brinkman, R. R., et al. Human monogenic disorders—A source of novel drug targets. Nature Reviews Genetics 7, 249–260 (2006) (link to article)
Huntington's Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell 72, 971–983 (1993)
Lowry, W. E., et al. Generation of human induced pluripotent stem cells from dermal fibroblasts. Proceedings of the National Academy of Sciences 105, 2883–2888 (2008)
O'Connor, T. P., & Crystal, R. G. Genetic medicines: Treatment strategies for hereditary disorders. Nature Reviews Genetics 7, 261–276 (2006) (link to article)
Online Mendelian Inheritance in Man, http://www.ncbi.nlm.nih.gov/omim/ (accessed August 18, 2008)
Park, I. H., et al. Disease-specific induced pluripotent stem cells. Cell 134, 1–10 (2008a)
Park, I. H., et al. Reprogramming of human somatic cells to pluripotency with defined factors. Nature 451, 141–146 (2008b)
Platt, O. S., et al. Hydroxyurea enhances fetal hemoglobin production in sickle cell anemia. Journal of Clinical Investigation 74, 652–656 (1984)
Takahashi, K., et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131, 861–872 (2007)
Weatherall, D. J. Pharmacological treatment of monogenic disease. Pharmacogenomics Journal 3, 264–266 (2003)
Yu, J., et al. Induced pluripotent stem cell lines derived from human somatic cells. Science 318, 1917–1920 (2007)










