


 Figure 1
Figure 1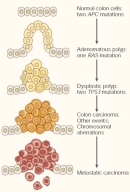




















« Prev Next »
According to the American Cancer Society, an estimated 294,120 men and 271,510 women will die of cancer in the United States in the year 2008. Table 1 shows the predicted distribution and percentages of these deaths (American Cancer Society, 2008).
Table 1: Predicted Distribution of U.S. Cancer Deaths in 2008
|
Males: 294,120 Cancer-Related Deaths |
Females: 271,530 Cancer-Related Deaths |
||
| Lung and bronchus | 31% | Lung and bronchus | 26% |
| Prostate | 10% | Breast | 15% |
| Colon and rectum | 8% | Colon and rectum | 9% |
| Pancreas | 6% | Pancreas | 6% |
| Liver and bile duct | 4% | Ovary | 6% |
| Leukemia | 4% | Non-Hodgkin's lymphoma | 3% |
| Esophagus | 4% | Leukemia | 3% |
| Urinary bladder | 3% | Uterine corpus | 3% |
| Non-Hodgkin's lymphoma | 3% | Liver and bile duct | 2% |
| Kidney and renal pelvis | 3% | Brain | 2% |
| All other sites | 24% | All other sites | 25% |
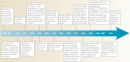
Unfortunately, these high numbers are nothing new. For example, in 2005, cancer was a leading cause of death in the United States, second only to heart disease and representing 22.8% of the nation's deaths. Moreover, despite impressive declines in other major forms of chronic illness, including heart disease and cerebrovascular disease, from 1950 to 2005, the number of cancer-related deaths showed only a slight decline during this same period. Currently, statistics indicate that during their lifetime, one in two men and one in three women will develop cancer. Thus, since 1971, when President Nixon declared "war" on cancer, the efforts of many researchers have focused on the early detection, prevention, and targeted destruction of this prevalent and devastating condition (Figure 1).
Today, scientists are armed with an entirely new arsenal of weapons in their fight against cancer: the human genome has been sequenced in its entirety, a large collection of genes have been identified in which mutations contribute to cancer, and an ever-expanding array of technologies is being used to identify new cancer-associated genes. Genetic testing can be employed to determine inherited cancer risk, or to obtain a genetic "fingerprint" of a tumor. In the future, an increasing number of cancer therapies will be genetically tailored to specifically and effectively target cancer cells. However, in order to understand the basis of these treatments, it is first necessary to understand the general genetic mechanisms by which cancer arises.
Cancer Is a Multistep Process
Cancer refers to a group of diseases that share a common overall phenotype: uncontrollable cell growth and proliferation (Balmain, 2001; Futreal et al., 2004). Cancer is a multistep process during which cells acquire a series of mutations that eventually lead to unrestrained cell growth and division, inhibition of cell differentiation, and evasion of cell death. As cancerous tumors grow in size, they stimulate a process called angiogenesis, during which they acquire a new source of blood vessels that provides them with oxygen and nutrients. Eventually, tumor cells invade the surrounding tissue and spread to other parts of the body in a process called metastasis (Figure 2).
Cancer is primarily a genetic disease that is clonal in nature. It usually starts when a single cell acquires a series of mutations, usually one after another, that collectively change a once-normal cell into a cancerous cell that divides uncontrollably and may eventually spread throughout the body. Gene mutations associated with cancer can be passed on through the germ line (i.e., inherited from one's parents), or they can be acquired through somatic mutations. It takes time for a cell to accumulate all of the mutations required to become cancerous, which is why cancer risk often increases with age.
Progression to cancer can occur more rapidly if an individual inherits a germ-line mutation in a cancer-associated gene; this person's cells will be one step closer to cancer from the very beginning. According to the Cancer Gene Census (Wellcome Trust Sanger Institute 2008), among the currently known cancer genes, 70 are associated with germ-line mutations, and 342 are associated with somatic mutations. Furthermore, 281 cancer-associated chromosomal translocations have also been identified.
Genes That Drive Cancer: Tumor Suppressor Genes and Proto-oncogenes
Mutations in two general types of genes lead to cancer: tumor suppressor genes, which normally act like "brakes" to inhibit cell growth and division, and proto-oncogenes, which normally act like "gas pedals" to accelerate cell growth and division. Mutations that inhibit the activity of tumor suppressor genes or that overactivate proto-oncogenes can drive cells to cancer; in either case, cells lose their brakes, hit the gas pedal, and accelerate uncontrollably toward a cancerous state.
Tumor suppressor genes were first discovered by Alfred Knudson through his studies of the inheritance of retinoblastoma, a childhood form of retinal cancer (Knudson, 1971). Mutations that inactivate tumor suppressor genes (called loss-of-function mutations) can be caused by deletion of the tumor suppressor gene, by a point mutation that leads to a functionally inactive tumor suppressor gene-encoded protein, or by a mutation that disrupts the promoter region of the tumor suppressor gene, leading to a loss or reduction in its expression. In most cases, tumor suppressor gene loss-of-function mutations are recessive, meaning that both copies of a cell's tumor suppressor gene must be mutated in order for cancer to occur.
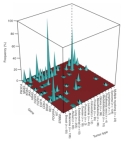
A bladder cancer-associated mutation in the human HRAS gene, which converts the twelfth glycine residue to a valine in the HRAS-encoded protein, was the first somatic mutation in a proto-oncogene shown to cause cancer (Reddy et al., 1982). Mutations in proto-oncogenes that convert them to oncogenes (called gain-of-function mutations) can be caused by single base-pair changes that lead to a hyperactive form of the proto-oncogene, by a gene amplification event that results in many extra copies of the proto-oncogene within the genome and higher expression levels of that gene, or by a mutation in the promoter region of the proto-oncogene that leads to an increase in its expression. Within the cell, these mutations behave in a dominant fashion, meaning that one changed allele can lead to uncontrolled cell proliferation.A recent high-throughput study shows that some types of cancer are associated with mutations in a specific proto-oncogene or in a specific pair of proto-oncogenes (Thomas et al., 2007). In this study, researchers screened 1,000 different tumor samples representing 17 different forms of cancer; the samples included individual tumor samples, primary tumor specimens, cancer cell lines, short-term cultures of tumor samples, and xenografts derived from tumor cell lines. The researchers screened 238 known oncogenic mutations in 17 different proto-oncogenes. Out of the 17 genes, 14 were mutated in at least one sample, and 34% of the 238 oncogenic mutations were detected. Thirty percent of the tumor samples contained at least one oncogenic mutation.
Figure 3 shows the association between certain forms of cancer and specific oncogenic mutations. Figure 4a shows the correlation between the various oncogenic mutations among the genes examined and the different types of tumors screened. Black bars indicate the occurrence of oncogenic mutations, and red bars indicate oncogenic mutations that occurred in pairs. Researchers found that 30% of all mutations in PIK3CA occurred in combination with a second oncogenic mutation (usually in KRAS), and that oncogenic mutations in the BRAF gene often occurred in combination with a mutation in the HRAS gene (see Figures 4b and 4c, respectively). As the researchers screened for known mutations in the 17 different proto-oncogenes, they also identified several previously unknown oncogenic mutations. The results of this study show that mutations in this set of 17 proto-oncogenes have a high propensity to lead to cancer, and that several paths can lead to cancer.
Boveri’s Early Predictions
In 1914, Theodor Boveri published a manuscript in which he put forth his hypothesis that the abnormal growth characteristics of tumor cells might be due to abnormal numbers of chromosomes within the tumor cells (Boveri, 1914), a phenomenon called aneuploidy. Boveri was studying chromosome segregation in fertilized sea urchin eggs and found that when a single egg was fertilized by two sperm cells, the resulting cell underwent abnormal development and/or cell death when the chromosome number was severely imbalanced. Boveri recognized the important connection between abnormal chromosome number and abnormal cell growth and division, which he noted was likely to be relevant to cancer (Balmain, 2001). Based on his observations, Boveri made several predictions that, in retrospect, are simply stunning (Figure 5). Indeed, a shared feature of nearly every tumor cell is its propensity to maintain an abnormal number of chromosomes, a property known as genetic instability.
The Road Map to Cancer
Cells may take the scenic route or the freeway to cancer, but most will acquire a common set of tumorigenic capacities along the way. Cancer geneticists have identified a set of distinct physiological changes that cells must undergo on their way to becoming cancerous (Hanahan & Weinberg, 2000). Under normal conditions, cells divide in response to growth factor signals, and cells cease dividing in response to anti-growth signals. The signals that moderate cell division are often released by neighboring cells. In response to genetic or environmental insult, normal cells will undergo programmed cell death or apoptosis, a highly-regulated process during which cells die before problems can arise. Chromosome ends, called telomeres, are designed in such a way that they become shorter and shorter every time a cell divides; a special enzyme called telomerase acts on telomeres to extend their length, but this enzyme is not active in most normal cells. Therefore, most normal cells can only divide a certain number of times before their telomeres shorten to the extent that critical genetic information is lost.
In order to become cancerous, normal cells must override the control processes that regulate cell growth and cell division. This ability to override normal control mechanisms is highlighted in the six so-called "hallmarks" of cancer:
(1) the ability to divide in the absence of growth factor stimulation, (2) the ability to divide in the presence of anti-growth signals, (3) the inability to undergo apoptosis, (4) the ability to maintain telomere length despite repeated cell divisions, (5) stimulation of angiogenesis, and (6) the ability to invade surrounding tissues and metastasize to other parts of the body (Figure 6; Hanahan & Weinberg, 2000).Although cells must acquire each of these capacities in order to become cancerous, they can take different routes to reach the final destination. In other words, they may pass through the six steps in any order, although the ability to metastasize is typically one of the last steps (Figure 7). Furthermore, the route to cancer may be shorter for individuals who have inherited a germ-line mutation in a cancer-associated gene from one or both of their parents. In the end, not all of the cells within a tumor will acquire the same set of subsequent mutations, which means that all tumors represent genetically heterogeneous groups of cells.
Today’s War on Cancer: Using Genes to Tailor Therapy
Cancer progression involves a complex series of genetic changes that occur over time. Genetic screening using blood samples can be used to identify germ-line mutations in individuals with a family history of cancer in order to assess cancer risk. In addition, genetic analysis of a tumor can be used to determine the somatic changes that have occurred. Knowledge of both germ-line mutations and tumor-associated somatic mutations can in turn be used to tailor cancer therapy in order to most effectively and specifically target tumor cells. Thus, although tremendous progress has been made since the "war" on cancer was declared in 1971, this war is still far from over.
References and Recommended Reading
American Cancer Society. Cancer Facts and Figures 2014.
Balmain, A. Cancer genetics: From Boveri and Mendel to microarrays. Nature Reviews Cancer 1, 77-82 (2001) doi:10.1038/35094086 (link to article)
Boveri, T. In Zur Frage der Entstehung Maligner Tumoren (Jena, Gustav Fisher, 1914)
Futreal, P. A., et al. A census of human cancer genes. Nature Reviews Cancer 4, 177–183 (2004) doi:10.1038/nrc1299 (link to article)
Hanahan, D., & Weinberg, R. A. The hallmarks of cancer. Cell 100, 57–70 (2000)
Knudson, A. G., Jr. Mutation and cancer: Statistical study of retinoblastoma. Proceedings of the National Academy of Sciences 68, 820–823 (1971)
Reddy, E. P., et al. A point mutation is responsible for the acquisition of transforming properties by the T24 human bladder carcinoma oncogene. Nature 300, 149-152 (1982) doi:10.1038/300149a0 (link to article)
Thomas, R. K., et al. High-throughput oncogene mutation profiling in human cancer. Nature Genetics 39, 347–351 (2007) doi:10.1038/ng1975 (link to article)
Wellcome Trust Sanger Institute. Cancer Genome Project: Cancer Gene Census. (2008)










