


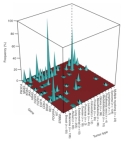
 Figure 1
Figure 1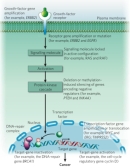















« Prev Next »
One out of every two men and one out of every three women will develop cancer during their lifetime (American Cancer Society, 2008). The current list of known cancer genes includes 70 genes associated with germline mutations and 342 genes associated with somatic mutations. Generally speaking, however, mutations in two basic classes of genes—proto-oncogenes and tumor suppressor genes—are what lead to cancer. In fact, a recent high-throughput study of proto-oncogene mutations in 1,000 different tumor samples representing 17 different types of cancer showed that mutations in a set of 14 proto-oncogenes are associated with a high propensity for cancer. (See Figure 1 for a depiction of the association between these genes and certain forms of cancer.) Moreover, this study also revealed that the 14 proto-oncogenes in question are associated with diverse cellular functions (Thomas et al., 2007). But what drives cancerous cells to grow and divide uncontrollably and escape cell death, and just how are proto-oncogenes involved in this process?
Introduction to Proto-oncogenes
Proto-oncogenes are a group of genes that cause normal cells to become cancerous when they are mutated (Adamson, 1987; Weinstein & Joe, 2006). Mutations in proto-oncogenes are typically dominant in nature, and the mutated version of a proto-oncogene is called an oncogene. Often, proto-oncogenes encode proteins that function to stimulate cell division, inhibit cell differentiation, and halt cell death. All of these processes are important for normal human development and for the maintenance of tissues and organs. Oncogenes, however, typically exhibit increased production of these proteins, thus leading to increased cell division, decreased cell differentiation, and inhibition of cell death; taken together, these phenotypes define cancer cells. Thus, oncogenes are currently a major molecular target for anti-cancer drug design.
Clues to Cancer from Viruses That Hijack Host DNA
Of course, viruses cause the common cold, the flu, and pneumonia, but they can also cause cancer. An American scientist named Peyton Rous discovered the first tumor-causing virus, called the Rous sarcoma virus, early in the twentieth century. Rous was studying the transmission of tumors in chickens. He found that he could induce tumor formation in a once-healthy chicken by injecting small pieces of a tumor taken from a cancer-prone chicken (Rous, 1910). Rous then made extracts from the chicken tumors and filtered them through small pores that blocked the passage of bacteria. Interestingly, he noted that these cell-free extracts also induced tumor formation in healthy chickens (Rous, 1911). Because the pore size of the filter was not small enough to exclude viruses, Rous concluded that a virus must be responsible for induction of tumor formation.
For his seminal discoveries, Rous won the Nobel Prize in 1966. In his Nobel lecture, Rous acknowledged the work of Ellerman and Bang, who discovered in 1908 that a virus-containing filtrate could transmit leukemia to chickens. Despite these findings, Ellerman and Bang's discovery was not widely recognized at the time because leukemia was not yet associated with cancer.
Rous was only the first in a long list of Nobel Prize-winning scientists who uncovered previously unknown links between the Rous sarcoma virus and cancer. For instance, the Rous sarcoma virus was later shown to be a retrovirus through a series of Nobel Prize-winning studies in the laboratories of David Baltimore, Howard Tenin, and Renato Dulbecco. In addition, in 1975, Nobel Prize winners Michael Bishop and Harold Varmus concluded that the Rous sarcoma viral gene responsible for causing cancer was in fact a host gene that had been hijacked by the virus. The host cell proto-oncogene was called c-src, and the Rous sarcoma viral oncogene was called v-src. Bishop and Varmus used different strains of Rous sarcoma virus in their research, and in all of the strains, they identified the v-src oncogene as responsible for causing cancer. These scientists started by generating a nucleic acid probe for use in hybridization experiments, and they found that the c-src gene was present in the genome of many species. They then showed that the host cell c-src gene was normally involved in the positive regulation of cell growth and cell division. Following infection, however, the v-src oncogene was expressed at high levels in the host cell, leading to uncontrolled host cell growth, unrestricted host cell division, and cancer.
Interestingly, although the Rous sarcoma virus itself is not capable of infecting human cells, humans have their own version of the c-src gene called SRC, and high levels of SRC expression are associated with several forms of human cancer.
From Good to Bad: How Proto-Oncogenes Become Oncogenes
Today, more than 40 different human proto-oncogenes are known. But what types of mutations convert these proto-oncogenes into oncogenes? The answer is simple: Oncogenes arise as a result of mutations that increase the expression level or activity of a proto-oncogene. Underlying genetic mechanisms associated with oncogene activation include the following:
- Point mutations, deletions, or insertions that lead to a hyperactive gene product
- Point mutations, deletions, or insertions in the promoter region of a proto-oncogene that lead to increased transcription
- Gene amplification events leading to extra chromosomal copies of a proto-oncogene
- Chromosomal translocation events that relocate a proto-oncogene to a new chromosomal site that leads to higher expression
- Chromosomal translocations that lead to a fusion between a proto-oncogene and a second gene, which produces a fusion protein with oncogenic activity
Examples of Oncogenes
Many proto-oncogenes play an important role during embryogenesis, because they are often involved in stimulating cell growth and proliferation as an organism develops. Some proto-oncogenes also negatively regulate cell differentiation. Proto-oncogene activities are typically turned off once the developmental processes they regulate are completed. However, if proto-oncogene activity remains high, or if proto-oncogenes are inappropriately reactivated later in life, cancer may occur.
A number of proto-oncogenes code for cell surface receptors that span the plasma membrane and bridge communication between the extracellular environment and the inside of the cell. These transmembrane receptors consist of three parts: an extracellular region that is exposed to the outside of the cell and acts like an antenna to collect outside signals; a transmembrane region that spans the plasma membrane; and an intracellular region that often has its own enzymatic activity and can associate with other proteins located inside the cell.
In order to grow and divide, cells respond to outside signals through the binding of extracellular ligands to the extracellular region of these transmembrane receptors (Figure 2). Often, these ligands are growth factors that stimulate cell division and growth, or angiogenic factors that stimulate new blood vessel formation. When a ligand binds to a receptor, the receptor will frequently undergo a change in its conformation (shape), which in turn leads to activation of the intracellular domain and a chain of intracellular events that regulate cell growth, proliferation, angiogenesis, or death. Examples of proto-oncogenic receptors include EGFR, the receptor of the epidermal growth factor (EGF) that is involved in growth factor-mediated signaling, and KDR, the receptor of the vascular endothelial growth factor (VEGF) that is involved in angiogenesis.
Proto-oncogenes can also code for intracellular proteins that normally act downstream of cell surface receptor pathways to stimulate cell growth and division. Examples of these downstream signaling proteins include HRAS and KRAS. Additionally, some proto-oncogenes, including cyclin D1 (CCND1) and cyclin E1 (CCNE1), normally act to push cells through distinct stages of the cell cycle when the cells receive the appropriate signals. When these proto-oncogenes are expressed at higher than normal levels, or when their expression is inappropriately turned on, cancer can occur.
Oncogene activation can also arise through chromosomal translocation events. The Philadelphia chromosome, discovered in 1960 in the Philadelphia laboratories of Peter Nowell and David Hungerford, is the best-known example of an oncogenic chromosomal translocation (Nowell & Hungerford, 1960). In this case, one end of chromosome 9 is exchanged with one end of chromosome 22. At the broken end of chromosome 22 lies the BCR gene, which fuses with a fragment of chromosome 9 that carries the ABL1 gene; this fused chromosome is called the Philadelphia chromosome. When the chromosome ends fuse, the two genes also fuse with each other to become BCR-ABL (Heisterkamp et al., 1985). The fused gene is expressed, and it encodes a protein that exhibits high protein tyrosine kinase activity, courtesy of the ABL1 half of the protein. The unregulated expression of this protein activates a repertoire of other proteins that are involved in cell cycle regulation and stimulation of cell division. As a result, the Philadelphia chromosome is associated with chronic myelogenous leukemia (CML) and several other forms of leukemia.
Targeting Oncogene Addiction to Treat Cancer

© 2008 Nature Publishing Group Weinstein, I. B. et. al., Mechanisms of Disease: oncogene addiction a rationale for molecular targeting in cancer therapy, Nature Clinical Practice Oncology >b>3, 448-457 (2006). All rights reserved. 
© 2006 Nature Publishing Group Weinstein, I. B. et al. Mechanisms of Disease: oncogene addiction - a rationale for molecular targeting in cancer therapy. Nature Clinical Practice Oncology 3, 449 (2006). All rights reserved.
Some forms of cancer occur early in life and are associated with mutations in a single gene. For example, retinoblastoma, which is a childhood form of retinal cancer, is caused by mutations in the RB1 gene, which is a tumor suppressor gene. However, cancer is more often a multistep process during which cells acquire a series of mutations that collectively lead to decreased tumor suppressor gene function and increased proto-oncogene function. The net result is a heterogeneous tumor cell population that grows uncontrollably, divides without restraint, and fails to respond to signals that would normally cause cell death.
Cancer cells nearly always contain abnormal numbers of chromosomes, a phenomenon called aneuploidy, which leads to altered dosage and expression of all of the genes carried on a given chromosome. As cancer cells rapidly grow and divide, they do not cope well with extra chromosomes, which leads to abnormal chromosome segregation and additional changes in chromosome number as the cells continue to divide. This well-established property of cancer cells is termed genetic instability.
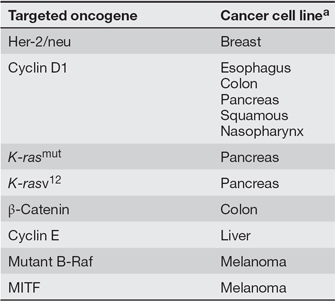
If cancer cells undergo so many different changes, how could we possibly hit all of the different targets required to inhibit their growth and proliferation? Researchers now believe that cancer cells may rely more heavily on certain oncogenic mutations than others for their growth, proliferation, and survival. This concept has been termed oncogene addiction (Weinstein & Joe, 2006). Therefore, by taking aim at the activity of a specific oncogene known to play a critical role in a certain type of cancer, researchers believe that they may be able to effectively target cancer cells, despite the presence of other mutations and an ever-changing number of chromosomes. Table 1 provides a list of human oncogenes that are associated with oncogene addiction. Inhibition of these genes' expression in human cancer cell lines leads to growth inhibition, a decreased ability to form tumors, and, in some cases, an increased sensitivity to chemotherapeutic agents. Table 2 provides a list of human oncogenes that can be targeted by drugs (agents) with or without chemotherapy (combination or monotherapy, respectively) in certain forms of cancer (disease).
As discussed earlier, CML is associated with a specific chromosomal translocation event that leads to the expression of a fusion protein consisting of BCR and ABL. Evidence that cells expressing the BCR-ABL fusion protein suffer from oncogene addiction comes from studies using imatinib, an ABL kinase inhibitor, to treat CML patients (Druker, 2002). This drug, which targets the tyrosine kinase activity of ABL, has been one of the biggest success stories in cancer treatment over the last decade. The ability to successfully treat CML patients with a drug that targets a single oncogenic target provides strong support for the theory of oncogene addiction. However, more long-term clinical studies using imatinib to treat CML patients are showing that some patients may begin to show signs of drug resistance over time.
Today, academic researchers, biotechnology companies, and pharmaceutical companies are continuing to develop approaches for targeting oncogene activity in the ongoing war on cancer (Chin & Gray, 2008). The approaches taken include using agents that bind and inhibit receptor activity, small RNA molecules that target oncogene expression, and drugs that inhibit the activity of downstream signaling proteins. The ability of cancer cells to evolve rapidly, combined with the heterogeneous nature of cancer cell populations, will continue to challenge researchers in years to come. Thus, like cancer cells, our approach to cancer therapy must also continue to evolve.
References and Recommended Reading
Adamson, E. D. Oncogenes in development. Development 99, 449–471 (1987)
American Cancer Society. Cancer Facts and Figures 2008.
Chin, L., & Gray, J. W. Translating insights from the cancer genome into clinical practice. Nature 452, 553–563 (2008) doi:10.1038/nature06914 (link to article)
Druker, B. J. Perspectives on the development of a molecularly targeted agent. Cancer Cell 1, 31–36 (2002)
Ellerman, C., & Bang, O. Experimentelle Leukämie bei Hühnern. Zentralblatt fur Bakteriologie 46, 595–609 (1908)
Heisterkamp, N., et al. Structural organization of the bcr gene and its role in the Ph' translocation. Nature 315, 758–761 (1985) doi:10.1038/315758a0 (link to article)
Nowell, P. C., & Hungerford, D. A. A minute chromosome in human chronic granuloytic leukemia. Science 142, 1497 (1960)
Rous, P. Transmissible avian neoplasm (Sarcoma of the common fowl). Journal of Experimental Medicine 12, 696–705 (1910)
———. Transmission of a malignant new growth by means of a cell-free filtrate. Journal of Experimental Medicine 13, 397-411 (1911)
Thomas, R. K., et al. High-throughput oncogene mutation profiling in human cancer. Nature Genetics 39, 347–351 (2007) (link to article)
Weinstein, I. B., & Joe, A. K. Mechanisms of disease: Oncogene addiction—a rationale for molecular targeting in cancer therapy. Nature Clinical Practice Oncology 3, 448–457 (2006) (link to article)










