
Abstract
Hypomyelinating leukodystrophy (HLD) is a rare genetic heterogeneous disease that can affect myelin development in the central nervous system. This study aims to analyze the clinical phenotype and genetic function of a family with HLD-7 caused by POLR3A mutation. The proband (IV6) in this family mainly showed progressive cognitive decline, dentin dysplasia, and hypogonadotropic hypogonadism. Her three old brothers (IV1, IV2, and IV4) also had different degrees of ataxia, dystonia, or dysarthria besides the aforementioned manifestations. Their brain magnetic resonance imaging showed bilateral periventricular white matter atrophy, brain atrophy, and corpus callosum atrophy and thinning. The proband and her two living brothers (IV2 and IV4) were detected to carry a homozygous mutation of the POLR3A (NM_007055.4) gene c. 2300G > T (p.Cys767Phe), and her consanguineous married parents (III1 and III2) were p.Cys767Phe heterozygous carriers. In the constructed POLR3A wild-type and p.Cys767Phe mutant cells, it was seen that overexpression of wild-type POLR3A protein significantly enhanced Pol III transcription of 5S rRNA and tRNA Leu-CAA. However, although the mutant POLR3A protein overexpression was increased compared to the wild-type protein overexpression, it did not show the expected further enhancement of Pol III function. On the contrary, Pol III transcription function was frustrated (POLR3A, BC200, and tRNA Leu-CAA expression decreased), and MBP and 18S rRNA expressions were decreased. This study indicates that the POLR3A p.Cys767Phe variant caused increased expression of mutated POLR3A protein and abnormal expression of Pol III transcripts, and the mutant POLR3A protein function was abnormal.
Similar content being viewed by others

Introduction
Leukodystrophy is a group of heterogeneous neurodegenerative diseases characterized by abnormal white matter imaging1. They are classified as hypomyelinating leukodystrophy (HLD) and non-hypomyelinating leukodystrophy based on white matter abnormalities on magnetic resonance imaging (MRI) of the brain2. POLR3-related hypomyelinating leukodystrophy (POLR3-HLD) is an autosomal recessive hypomyelinating leukodystrophy featured by neurological and non-neurological systems, and its typical clinical phenotype includes hypomyelination, hypodontia, and hypogonadotropic hypogonadism, therefore it is also known as 4H syndrome3. POLR3-HLD is caused by mutations in the POLR3A, POLR3B, POLR1C, and POLR3K genes, which encode subunits of human RNA polymerase III (Pol III)4. Pol III is the largest of the three RNA polymerases, containing 17 subunits. POLR3A encodes the largest subunit of Pol III, which together with POLR3B forms the catalytic center of the enzyme. Pol III is involved in the transcription of a variety of small molecules of non-coding RNAs (NC-RNAs), including transfer RNAs (tRNAs), 5S ribosomal RNA (rRNA), 7SL and 7SK RNAs, and U6 small nuclear RNA5. Most Pol III transcripts are involved in the regulation of essential processes such as transcriptional regulation, RNA processing, and protein translation6, and the normal function of Pol III plays a key role in the maintenance and development of myelin7. In addition, Pol III-transcribed brain cytoplasmic 200 RNA (BC200 RNA) is highly expressed in the central nervous system (CNS), so its role in neurological disorders deserves further investigation. BC200 RNA is a primate brain-specific non-messenger RNA8, which is considered to be a translational regulator in the synaptic-dendritic structural domain of neurons and is associated with the regulation of dendritic local protein synthesis9. However, the role of BC200 RNA in myelin formation is rarely studied.
The impairment of Pol III or tRNA function can easily affect the CNS. A variety of neurological diseases, including several leukodystrophy, have been found to result from mutations in genes associated with tRNA biology10,11,12,13. POLR3A gene is constitutively expressed in all tissues, and although it is not expressed at very high levels, it is the highest in the cerebellum14. POLR3A mutation may lead to dysfunction of Pol III, which affects the expression of certain tRNAs that are more abundant in the CNS, and thus disrupts protein synthesis15. In our previous work, we identified a hypomyelinating leukodystrophy-7 (HLD-7) family and detected the homozygous variation of POLR3A (NM_007055.4) gene c. 2300G > T (p.Cys767Phe). At present, there is no literature to study the function of the mutation site, and its clinical significance is unclear. Therefore, this study intends to analyze the clinical phenotype of the family and study the function of the mutation site to further investigate the pathogenicity of this mutation. It is expected that this study can improve clinicians’ understanding of the HLD-7 and POLR3-HLD disease spectrum and provide a reference for clinical diagnosis and treatment of this disease.
Materials and methods
Research object
The proband (IV6, Fig. 1a), a 36-year-old female, presented to our hospital with the chief complaint of “dizziness and memory decline for 16 years”. Collect the relevant medical history and clinical data of the proband and other members of the family, such as biochemistry, serum ceruloplasmin, thyroid function including thyroid stimulating hormone (TSH), free triiodothyronine (FT3), and free thyroxine (FT4), parathyroid hormone, and sex hormone (follicle-stimulating hormone [FSH], luteinizing hormone [LH], estradiol [E2], progesterone [P4], testosterone [T], prolactin [PRL]). The results of breast color ultrasound, vaginal color ultrasound, brain MRI, electroencephalogram (EEG), electromyogram (EMG), blink reflex, somatosensory evoked potential (SEP), and event-related evoked potential P300 of the proband were collected. This study was approved by the Ethics Committee of Fujian Provincial Hospital, China, and all the family members participating in this study signed informed consent forms.

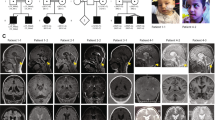
Genogram and Sanger sequencing diagram of the family. (a) Pedigree diagram of a family with hypomyelinating leukodystrophy-7, in which III1 is also complicated by Finnish type renal amyloidosis; the proband (arrow) carries a homozygous mutation of POLR3A (NM_007055.4) c.2300G > T (p.Cys767Phe); blue indicates carrying POLR3A (NM_007055.4) c.2300G > T (p.Cys767Phe) variant. (b) The proband had dentin dysplasia. (c) c.2300G > T (p.Cys767Phe) homozygous mutant at exon 17 of POLR3A gene. (d) c.2300G > T (p.Cys767Phe) heterozygous mutant at exon 17 of POLR3A gene. (e) Sanger sequencing verification map of the wild-type POLR3A gene.
Extraction of genomic DNA
Peripheral blood of the proband and family members were collected with an ethylenediaminetetraacetic acid anticoagulant tube, and genomic DNA of the proband and family members were extracted according to the instructions of the QIAamp DNA Blood Mini Kit (QIAGEN Co., Ltd.). The extracted DNA samples were examined using the NanoDrop instrument, agarose gel electrophoresis, and Qubit, and used for the subsequent sequencing process.
Whole-exome sequencing and variant screening and interpretation
Next-generation sequencing (NGS) was performed on the proband. Break genomic DNA and use a reagent kit for end-repaired, adding adenine bases, and connecting connectors, the genomic DNA library was obtained after polymerase chain reaction (PCR) amplification and PCR product purification. NEXome Core Panel and Hybridization and Wash kits were used for a whole-exome capture in the library, and the quality of the library was tested after PCR linear amplification. The coverage of the target area is 99.63%, and the average sequencing depth of the target area is 160.88 ×. After passing the standard, PE 150 sequencing was performed using a DNBSEQ-T7 sequencer. The capture target genes involved include NOTCH3, POLR3A, POLR3B, POLR3K, POLR1C, CYP7B1, and MADD. The sequenced fragments were compared with the UCSC hg19 human reference genome by BWA software, and the data were sorted and de-duplicated using Picard and Samtools markdup software, then the data were analyzed for single nucleotide variant and insertion and deletion using GATK software, and copy number variation at the exon level was detected using ExomeDepth. Combined with the data of mutation detection, the phenotypic-related sites were analyzed by Exomiser software, and the suspicious mutations were screened and interpreted according to the American College of Medical Genetics and Genomics guidelines. Primer 5.0 was used to design specific primers for the upstream and downstream positions of the target mutation site sequence, and the target region was amplified. Sanger sequencing was performed using an ABI 3730XL DNA sequencer (ABL, USA). The amplified fragment length of the target sequence of c. 2300G > T (p.Cys767Phe) of POLR3A (NM_007055.4) was 800 bp, and the primer was F: TGAGGGCCTCGGTATCTACAA; R: TGATCGGAAATCGGGCATCT. The annealing temperature was 60 °C. The primers were synthesized by Beijing liuhe BGI Technology Co., Ltd.
Plasmid construction and identification
The coding sequences (CDS) of POLR3A (NM_007055.4) and POLR3A p.Cys767Phe mutant were constructed into pCDH-CMV-MCS-EF1-copGFP-T2A-puro vector, XbaI/NotI was selected as the double digestion site, and the N-terminal end of the CDS carried the FLAG tag. The target gene was amplified and verified by sequencing. The plasmid vector construction and related PCR primer synthesis were done with the assistance of Wuhan gene create Co., Ltd (Wuhan, China).
Cell culture and transfection
The hela cells were placed in 1640 complete culture medium containing 1% Penicillin–Streptomycin Solution and 10% Fetal bovine serum and cultured at 37 °C in a 5% CO2 incubator, and the cells were routinely passaged at approximately 90% cell density. The hela cells were digested and collected by trypsin, and the cells were laid on a 6 cm cell-culture dish at a density of 5 × 105 cells/plate in an appropriate complete medium. The cells were incubated overnight in a 37 °C incubator with 5% CO2 according to the cell adherence, and transient transfection could be started after the cells were fully adherent. Prepare TurboFect-DNA Mix according to TurboFect instructions, add 6 µg of plasmid and 12 µL of TurboFect into 600 µL of Opti-Medium and mix gently, and then incubated at room temperature for 15 min. Add TurboFect-DNA Mix drop by drop to the cell culture medium of the above monolayer cells, shake the dish gently and mix well, and incubate in a 37 °C incubator containing 5% CO2. After 8–12 h of incubation, the medium was replaced with serum-containing medium preheated at 37 °C, and after continuing the culture up to 48 h, cell samples were collected for subsequent verification of cell function.
Real-time quantitative polymerase chain reaction (RT-qPCR)
The cells were collected for RNA extraction, reverse transcription, and qRT detection. Primers were designed with Primer 5.0 (Table 1).
Cell immunofluorescence
After being fixed, transparent, and blocked, the transfected hela cells were incubated overnight at 4 °C with the Flag primary antibody (Anti-FLAG 1:1000). The transfected hela cells were rinsed with phosphate-buffered saline (PBS) three times; the fluorescent secondary antibody (Goat Anti-Mouse, final concentration 2 μg/mL) was added and incubated at room temperature in the dark for 2 h, rinsed 3 times with PBS; stained with 4′,6-diamidino-2-phenylindole and incubated at room temperature for 10 min, rinsed 3 times with PBS; sealed with 50% glycerol; and photographed by laser confocal microscopy after sealing.
Western Blot
The hela cells were cultured and lysed, the total protein was extracted, and the protein concentration was detected with a multifunctional enzyme marking instrument. After the electrophoresis device was set up, protein samples, as well as protein Maker, were added to the wells on the electrophoresis gel in the desired order using a pipette or a loading needle. After electrophoresis, the proteins were wet transferred to the polyvinylidene fluoride (PVDF) membrane. The membrane after protein transfer was rinsed with Tris-buffered saline with 0.1% Tween® 20 (TBST) and sealed at room temperature for 1 h by adding 5% bovine serum albumin (BSA) blocking solution per 0.1 mL/cm2. After sealing, the PVDF membrane was rinsed with TBST once. The primary antibody (Anti-FLAG, 1:2000; Anti-α-Tubulin, 1:1000) was diluted with 5% BSA to the appropriate concentration, put into PVDF membrane and react at 37 °C for 1 h or overnight at 4 °C; after washing the membrane 3 times, add the secondary antibody (Goat Anti-Mouse) labeled with horseradish peroxidase diluted with 5% BSA, and put it into the PVDF membrane and react at room temperature for 1 h; wash for 5 times and then expose.
Statistics
The data of 2−△△CT values obtained by RT-qPCR were statistically analyzed by GraphPad Prism version 6.0 software. The measurement data were expressed as mean ± standard error of the mean. A one-way analysis of variance was used for comparison between multiple groups. The least significant difference test was used for comparison among groups.
Ethical approval
All procedures were performed in accordance with the tenets of the Declaration of Helsinki and the study was approved by the Ethics Committee of Fujian Provincial Hospital, Fuzhou, China. All participants and legal guardians of the minors involved in the present study provided written informed consent.
Results
Clinical phenotypes
The proband (IV6) is a 36-year-old woman. The proband had paroxysmal dizziness and memory decline at the age of 20, manifested by frequent forgetfulness, accompanied by rapid temper, but her life and work had not been significantly affected. Brain MRI in the local hospital showed white matter degeneration and cerebellar atrophy. Urine routine indicates microalbuminuria. No headache, limb numbness, fever, nausea, vomiting, speech confusion, and no dyskinesia. There was a history of delayed eruption of deciduous teeth and infertility. She has no history of drinking, taking drugs, and taking phenothiazines, no history of psychosis, encephalitis, brain trauma, and stroke, and no history of adverse reactions to vaccines. The proband had menarche at the age of 17 and had irregular menstruation, requiring long-term oral estradiol tablets/estradioland dydrogesterone tablets. Married at the age of 34, currently infertile. Her parents (III1 and III 2) were consanguineous marriage and had no similar medical history, but her three older brothers (IV1, IV2, and IV4) all had a history of cognitive dysfunction. Physical examination: height 167 cm, weight 60 kg, BMI 21.26 kg/m2, good nutritional status, normal blood pressure in both prone and standing positions, no K-F ring in the cornea, dentin dysplasia (Fig. 1b), no significant abnormalities in heart, lung, and abdominal examination. Breast and pubes development stagnated at Tanner Stage II, vulvar development was immature, and there were no deformities in spine and limb development. Nervous system physical examination: clear consciousness, clear speech, answering questions correctly, cooperative examination; memory slightly decreased, MMSE 27 points, MoCA 15 points (bachelor degree). The sense of smell was normal, and the cranial nerve examination was normal. The muscle strength, muscle tension, and tendon reflex of the limbs were normal. Bilateral superficial and deep sensation existed symmetrically. The movement was flexible, and the gait was normal. The bilateral finger-nose test was accurate, the alternating movement test was coordinated, the heel-knee-tibia test was normal, and Romberg’s sign was negative. Bilateral pathological signs were negative. Auxiliary examination: Thyroid function examination in other hospitals showed that TSH 0.92 (0.27 ~ 4.20) uIU/ml, FT3 5.87 (3.1 ~ 6.8) pmol/L, and FT4 16.9 (12 ~ 22) pmol/L; anti-mullerian hormone 5.24 (0.777 ~ 5.24) ng/ml; repeated monitoring of sex hormone levels showed E2 13.7 ~ 26 (49 ~ 291) pg/ml, FSH 0.05 ~ 0.17 (1.79 ~ 5.12) mIU/ml, LH 0.01 (1.20 ~ 12.86) mIU/ml, PRL 2.06 ~ 3.7 (3.34 ~ 26.72) ng/ml, P4 0.29 ~ 0.55 (5.16 ~ 18.56) ng/ml, and T 0.35 ~ 0.46 (0.1 ~ 0.75) ng/ml. At that time, the gonadotropin-releasing hormone (GnRH) stimulation test showed that LH and FSH had no significant response. Breast color ultrasound showed that the structure of the glandular layer was disordered and the arrangement was not clear. Vaginal ultrasound shows the posterior position of the uterus, with a clear contour and regular shape, approximately 42 mm × 39 mm × 27 mm in size; its muscular layer has uniform echoes and no obvious abnormal mass echoes was observed. After admission, a craniocerebral MRI showed multiple tiny nodular foci in the pituitary gland, considering microadenoma; hypomyelination of the white matter of the brain; brain atrophy; and atrophy and thinning of the corpus callosum (Fig. 2). EEG, EMG, and blink reflex tests showed no significant abnormalities (the detailed results of EMG can be found in Supplementary Table S1). SEP: both upper limbs were normal, the left lower limb was abnormal, and the right lower limb was normal (see Supplementary Table S2). P300 detection: P300 waveform differentiation and repeatability were acceptable, the latency was slightly prolonged and the reaction time was prolonged (Table 2). No obvious abnormality was found in biochemistry and serum ceruloplasmin.

Craniocerebral magnetic resonance imaging (MRI) results of the proband. (a–i) Craniocerebral MRI showed that the pituitary gland was approximately 3.8 mm in height, with a flat upper edge and a slightly sunken saddle base, and multiple tiny nodules with equal and slightly long T1 signals and slightly long T2 signals were seen inside, and the dynamic enhancement showed relatively slightly low signals, the larger one being approximately 2.2 mm in diameter; bilateral periventricular white matter atrophy and degeneration, with short linear and patchy equal and slightly long T1 and slightly long T2 signals, and slightly high T2-Flair signal; cerebral sulcus, cistern, and ventricle were generally widened; corpus callosum atrophy and thinning, particularly at the knee and body of the corpus callosum.
The proband’s eldest brother (IV1), height 180 cm, delayed puberty, unmarried and infertile. IV1 had delayed eruption of permanent teeth and lack of the right maxillary median incisor. Neurological symptoms appeared at the age of 16, mainly characterized by personality changes, emotional instability, memory loss, slurred speech, and progressive walking difficulties. He began to use a wheelchair at the age of 21 because he was unable to stand. He became bedridden around the age of 30 and has been dead for 3 years (age of death 38). The proband’s second brother (IV2) was 39 years old, height 184 cm, and had a son and a daughter. His children are all healthy. IV2 had delayed eruption of permanent teeth, delayed puberty, and no significant abnormalities in sex steroid levels. At the age of 20, he began to experience personality changes, manifesting as euphoria, exaggerated delusions, and gradually developing memory loss and unstable walking, and currently can still walk independently. Physical examination: there was no obvious abnormality in the general examination and physical examination of the heart, chest, and abdomen. He was clearly conscious, could answer correctly, and was more euphoric, but was uncooperative in the intelligence examination and could not complete the clock drawing test. The cranial nerve examination was negative, the muscle tension of the extremities was normal, the muscle strength of both upper limbs was grade 5, and the muscle strength of both lower limbs was grade 4. Mild atrophy of extremity muscles. The bilateral finger-nose test was normal, the heel-knee-tibia test was uncooperative, the walking straight line was not possible, Romberg’s sign was positive, and bilateral superficial and deep sensory examination was uncooperative. Tendon reflexes of both upper limbs (+), tendon reflexes of both lower limbs (+ + +). Patellar clonus (−), bilateral ankle clonus (+), and bilateral pathological signs (+). P300 detection: P300 waveform differentiation and repeatability were poor, with prolonged latency and reaction time (Table 2). The proband’s third brother (IV4), 38 years old, height 180 cm, and had 2 sons. His sons were both healthy. IV4 had delayed eruption of deciduous teeth and lack of the left maxillary canine; puberty was delayed but sex steroid levels were not significantly abnormal. He started to experience neurological symptoms at the age of 23, first manifesting as excessive speech and was treated with oral olanzapine in an external psychiatric unit (details unknown). Thereafter, he gradually developed decreased speech, dullness, and memory loss. He developed progressive unsteadiness in walking at the age of 30 and started using a wheelchair at the age of 35 due to inability to stand. Physical examination: partial cooperation on physical examination, malnourished appearance, and sent to hospital in a wheelchair. Cardiac, thoracic, and abdominal examinations showed no abnormalities. He was clearly conscious, with a dull expression and markedly decreased reaction speed, and was unable to complete the clock drawing test. Difficulty in eye tracking and scanning, with bilateral nasolabial folds symmetrical in a natural state, and tongue extension in the center; minimal speech, dysarthria, and the remaining cranial nerve examination were uncooperative. The muscle tension of both lower limbs was increased, and the muscle strength of the extremities was grade 3. Moderate-to-severe atrophy of extremity muscles. The ataxic motor and bilateral superficial and deep sensory examinations were uncooperative. Tendon reflexes of both upper limbs (+), tendon reflexes of both lower limbs (+ + +), patellar clonus (−), bilateral ankle clonus (+), and bilateral pathological signs (+). P300 detection: no definite P300 wave was observed. The craniocerebral MRI of the three old brothers of the proband all showed symmetrical white matter abnormalities around the ventricles, corpus callosum thinning, and cerebellar atrophy. Unfortunately, except for P300, IV2 and IV4 did not cooperate with EMG and SEP testing, so it was difficult to distinguish whether their extremity muscle atrophy was wasting atrophy or atrophy due to peripheral neuropathy. All members of this family also refused to undergo nerve biopsy. In addition, the father of the proband had manifestations of proteinuria and nephritis, and is currently undergoing hemodialysis in their local hospital due to uremia. He was diagnosed with Finnish type renal amyloidosis by gene sequencing in our department. The clinical features of the proband and family members were summarized in Table 2.
Screening for mutated genes
Through the analysis of NGS and exon capture techniques, a homozygous mutation c. 2300G > T (p.Cys767Phe) in exon 17 of POLR3A (NM_007055.4) was found in the proband (IV6), which led to the conversion of cysteine (Cys) to phenylalanine (Phe) at position 767 of POLR3A protein. The proband’s two surviving brothers (IV2 and IV4) also carried a homozygous mutation of POLR3A (p.Cys767Phe), and their consanguineous married parents (III1 and III2) carried a heterozygous mutation of POLR3A (p.Cys767Phe). In addition, V1, V2, V3, and V4 also carried the POLR3A (p.Cys767Phe) heterozygous mutation (Fig. 1c–e).
Cloning of POLR3A wild-type and p.Cys767Phe mutant
The pCDH-CMV- POLR3A-EF1-copGFP-T2A-Puro, pCDH-CMV- POLR3A c.2300G > T-EF1-copGFP-T2A-Puro, and control were transiently transfected. The fluorescence and growth of cells showed that the cells were in good condition (Fig. 3a).

The situation of cell culture and the detection results of Western Blot (WB). a. Basal culture growth of hela cells after transfection. b. The expression of exogenous POLR3A protein in the cells was detected by WB. Original blots/gels are presented in Supplementary Fig. S1.
Expression of POLR3A wild-type and p.Cys767Phe mutant in hela cells
The expression of exogenous POLR3A protein in the cells was detected by WB (Fig. 3b). The results showed that compared with the control group, the expression of POLR3A protein increased after overexpression of both POLR3A wild type and POLR3A c. 2300G > T mutants, and the expression of POLR3A protein was higher than that of the wild type after POLR3A c. 2300G > T mutation. The expression of POLR3A, MBP, BC200, 5S rRNA, tRNA Leu-CAA, 7SK RNA, 18S rRNA, 28S rRNA, and internal reference in cells was detected by RT-qPCR (Fig. 4). The results showed that the overexpression of wild-type POLR3A protein could significantly enhance Pol III transcription of 5S rRNA, tRNA Leu-CAA. However, although the mutant POLR3A protein overexpression was increased compared to the wild-type protein overexpression, it did not show the expected further enhancement of Pol III function. On the contrary, Pol III transcription function was frustrated (POLR3A, BC200, and tRNA Leu-CAA expression decreased), and the expression of MBP and 18S rRNA was decreased, suggesting that the mutant POLR3A protein function was abnormal.

Expression levels of Pol III transcripts. The relative expression of POLR3A, MBP, BC200, 5S rRNA, tRNA Leu-CAA, 7SK RNA, 18S rRNA, 28S rRNA, and internal reference were detected by Real-time quantitative polymerase chain reaction. Data are presented as mean ± standard error of the mean, n = 3; ns, not significant, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Localization of POLR3A wild-type and p.Cys767Phe mutant in cells
Immunofluorescence was used to detect the expression and localization of the POLR3A protein (Fig. 5). Considering the effect of endogenous POLR3A, this part of the experiment was performed using Flag antibody, and the results showed that wild-type POLR3A protein and POLR3A c.2300G > T mutant protein were uniformly distributed in the cytoplasm, and the expression of POLR3A protein was enhanced after POLR3A c. 2300G > T mutation.

The expression and localization of POLR3A protein in hela cells. The wild-type POLR3A protein and POLR3A c.2300G > T mutant protein were uniformly distributed in the cytoplasm, and the expression of POLR3A protein was enhanced after POLR3A c. 2300G > T mutation.
Bioinformatics prediction of proteins
The protein structure of POLR3A was searched at https://www.rcsb.org/sequence/7AE1, and the mutated protein structure was predicted by Chimera software. Using chimera software to predict the hydrogen bond of POLR3A protein structure, it can be seen that the original 767 Cys can form a hydrogen bond with 765 Ser and 764 Gly, but the hydrogen bond cannot be formed after mutation (Fig. 6).

Structure prediction of the POLR3A protein. a. Three-dimensional structure of POLR3A protein. b. The local base structure of Cys at position 767. c. The local base structure of mutation to 767 Phe. d. Local structure diagram of 767 Cys with adjacent amino acids. e. Local structure diagram of 767 Phe with adjacent amino acids. The black lines represent hydrogen bonds.
Discussion
HLD-7 is an autosomal recessive hereditary leukodystrophy associated with POLR3A mutation16. POLR3A is located on the q22.3 of chromosome 10 and consists of 31 exons, encoding Pol III catalytic subunit A (POLR3A) protein14. POLR3A is essential for normal nucleolus function, ribosome assembly, and protein translation17,18. POLR3A interacts with multiple subunits of Pol III. POLR3A pathogenic mutants can directly interfere with the ability to bind DNA, alter the catalytic cleft structure, and interfere with the protein interactions between POLR3A and other Pol III subunits, thereby impairing the normal assembly of Pol III19.
Interestingly, the effect of POLR3A mutations on protein expression seems to be variable in different studies. Previous studies have found that the level of POLR3A protein decreased in fibroblasts derived from patients with 4H syndrome, and the level of POLR3A protein in cortical and cerebral white matter of individuals with 4H syndrome decreased, with a more significant decrease in cerebral white matter15. Reduced levels of POLR3A protein were also observed in HEK293 clonal cell lines and MO3.13 cell lines carrying the POLR3A p.M852V mutation20. However, in a study of the effects of POLR3A mutations on mRNA and proteins in fibroblasts from patients with Wiedemann-Rautenstrauch syndrome, POLR3A mutations resulted in a decrease in wild-type POLR3A mRNA and POLR3A protein expression, while the expression of mutant proteins increased sharply 18. In addition, in the POLR3-HLD mouse model with the import of the POLR3A c.2015G > A (p.Gly672Glu) mutation, the levels of the POLR3A protein in the mutant mice and the major protein components of myelin in the mouse cerebellum (myelin basic protein (MBP), proteolipid protein, and myelin-associated glycoprotein) were comparable to those of wild-type mice, and the myelin formation of mutant mice was not significantly impaired 21. In fibroblasts from a patient carrying a compound heterozygous mutation in POLR3A c.3721G > A (p.Val1241Met) and the splicing region c.1771-6C > G, quantitative real-time PCR assays showed that the RNA expression level of Pol III target genes (HNRNPH2, UBB, LTF, and HSP90AA1) decreased and overexpression of wild-type POLR3A protein rescued the basic expression of Pol III target genes or reached higher levels, while overexpression of POLR3A protein with p.Val1241Met mutation did not rescue their expression or even appeared at lower levels16. This not only confirms the pathogenicity of POLR3A p.Val1241Met, but also suggests that the mutant POLR3A protein is functionally weaker than the wild type, and its elevated expression may even cause alteration or loss of Pol III function.
The transcriptional activity of Pol III is closely related to the regulation of cell growth, cell cycle, and cell differentiation processes22. Two hypotheses exist regarding potential possible mechanisms for POLR3-HLD: one is that mutations in the gene encoding the Pol III subunit result in reduced levels of tRNA and/or other NC-RNAs that are critical to myelin formation23. Oligodendrocytes are myelin-forming cells of the CNS. During myelin development or remyelination, oligodendrocytes need to produce large amounts of proteins to mature, and disruption of tRNA transcription can cause dysregulation of global protein translation during the critical phase of myelin development, ultimately leading to abnormal myelin formation5,24. The second hypothesis suggests that reduced Pol III function can lead to reduced levels of specific Pol III transcripts involved in transcription, RNA processing, and translation, thus affecting the development and function of oligodendrocytes and/or neurons23.
Alterations in the expression of POLR3A-regulated genes and ribosomal transcripts may affect ribosome production, protein synthesis, and the regulatory processes of RNA polymerases I and II18. 5S rRNA is the smallest RNA component of the ribosome, and the cytoplasmic ribosomes of all species contain highly conserved 5S rRNA25. The presence of 5S rRNA, a component of the large ribosomal subunit, is extremely important for the normal function of the ribosome26. 7SK RNA is a nuclear transcript produced by Pol III, which plays a crucial role in the transcriptional regulation of RNA polymerase II through binding to the transcriptional elongation factor P-TEFb27. It has been suggested that 7SK RNA is a major regulator of neuronal development and function28. Expression levels of tRNAimet, 5S rRNA, 7SK RNA, and 7SL RNA were found to be decreased in fibroblasts from patients with HLD affected by the POLR3K mutation26. In contrast, blood RNA analysis in patients with striatal involvement phenotype due to POLR3A mutation showed an overall decrease in tRNA abundance and an increase in 5S rRNA and 7SK RNA levels29. In a study in which endogenous POLR3A c.2554A > G (p.M852V) was introduced into HEK293 cells by CRISPR-Cas9 technique to obtain a homozygous mutant clone and two compound heterozygous clones with null allele, the POLR3A mRNA levels of the compound heterozygote with p.M852V mutation were decreased, but the POLR3A mRNA level of the homozygote with p.M852V mutation was not decreased; precursor tRNA levels were reduced as a whole in all three mutant cells20. In our study p.Cys767Phe mutant resulted in increased expression of mutant POLR3A protein. To further analyze the role of the p.Cys767Phe mutant in transcriptional control, we used RT-qPCR to detect the expression of Pol III transcripts. We observed that both tRNA-Leu-CAA and 18S rRNA levels were decreased after the POLR3A p.Cys767Phe mutation compared to the wild type. Considering that Pol III is central to tRNA production, changes in tRNA may be the main pathogenic mechanism of the POLR3A mutant15, and the decrease of 18S rRNA level suggests that this mutant may have an impact on the transcriptional regulation of RNA polymerase I. In addition, 5S rRNA levels were elevated after the p.Cys767Phe mutation compared to the wild type. Although the levels of the transcriptional regulator 7SK RNA after the p.Cys767Phe mutation were not significantly different from those of the wild type group, there was an overall trend of increased levels of 7SK RNA compared to the control group. The increased expression of Pol III-specific transcripts may be a compensatory response to the decrease of tRNA level and preferential recruitment of Pol III to their promoters29.
This study also observed that the POLR3A p.Cys767Phe mutation led to a decrease in the level of BC200 RNA. BC200 is a long non-coding RNA that plays an important regulatory role in the translation process of human neuronal cells30. BC200 RNA was the most downregulated Pol III transcript in several datasets of POLR3A p.M852V mutant cells; moreover, BC200 RNA was also decreased in fibroblasts from patients with POLR3-HLD and in the MO3.13 cell line of oligodendrocytes carrying the POLR3A c.2554A > G (p.M852V) mutation20. The above results indicated that BC200 RNA may be particularly sensitive to Pol III functional abnormalities20, and may play an essential role in the pathogenic mechanism of POLR3-HLD. However, considering that BC200 RNA was overexpressed in a variety of tumor cells and was critical to cancer cell survival and proliferation31, and MO3.13 cell lines were established from tumors, it cannot be excluded that BC200 RNA expression and function are unique to this cell line20,32. In addition, the POLR3A p.Cys767Phe mutation also caused the decrease of MBP mRNA expression in this study. MBP is the second most abundant protein in the myelin sheath of the CNS and is an adhesion agent in the formation of multilayered dense myelin sheaths of the mammalian CNS, playing an important role in myelin formation33. MO3.13 cell lines expressing oligodendrocyte precursor cells characteristics could differentiate into a more mature oligodendrocyte phenotype with enhanced MBP expression34. After the differentiation of MO3.13 cells, the mRNA level of the MBP gene in POLR3A p.M852V mutant cells was significantly lower than that in wild-type cells 20. This suggested that the POLR3A mutation could impair the expression of MBP, an important marker for mature oligodendrocytes. Indeed, the functional consequences of various mutations in the genes encoding Pol III subunits are unknown, and how Pol III mutations cause diseases that are mainly confined to the CNS and a few other tissues remains a mystery23. Because differences in the affected CNS cell types may result in different clinical phenotypes, Pol III dysfunction may affect different cell types differently, leading to a diversity of disease mechanisms23.
From a clinical perspective, there is also heterogeneity in the clinical phenotype caused by POLR3-HLD, which can involve both the neurological and non-neurological systems. Typical neurological involvement can present with neurodevelopmental delays such as learning difficulties and intellectual disability, and can also result in signs and symptoms of cerebellar, pyramidal, and extrapyramidal system involvement such as ataxia, tremor, dysarthria, spasticity, and dystonia35. Non-neurological features usually include dental abnormalities (absent or delayed eruption of teeth); endocrine abnormalities (delayed, halted, or absent puberty and short stature); and ocular abnormalities (myopia, optic atrophy, and cataracts)7. POLR3-HLD often occurs in infancy or childhood, and the earlier the age of onset, the more severe the clinical phenotype is likely to be36. Compared with POLR3B mutations, POLR3A mutations usually lead to more serious clinical phenotypes and shorter life expectancy for patients37. Craniocerebral MRI has high sensitivity in the diagnosis of white matter abnormalities in the CNS. The typical MRI presentation of POLR3-HLD was diffuse hypomyelination with variable T1 signal intensity (low, high, or equal signal), T2 high signal in the white matter of the brain, and relative T2 hypointense signal in specific structures such as the anterolateral nuclei of the thalami, dentate nucleus, globus pallidus, pyramidal tracts of the posterior limb of the internal capsule, and optic radiations; cerebellar atrophy; and thinning of the corpus callosum5,7,19,38. Compared to POLR3B mutations, hypomyelination was more obvious on MRI in patients with POLR3A mutations, whereas the changes in the cerebellar hemispheres and vermis were significantly milder35,39. POLR3A mutation can lead to gradually obvious cerebellar signs, but cases of cerebellar atrophy not observed by MRI have been previously reported40. It should be noted that diffuse hypomyelination is not a necessary imaging feature of POLR3-related disease, and patients with POLR3A or POLR3B mutations can present without hypomyelination41,42. However, the simultaneous appearance of the above MRI manifestations is a unique imaging feature of POLR3-HLD43.
Due to the high phenotypic heterogeneity of POLR3A mutations, different families carrying the same mutation may result in different phenotypes4. However, distinct intrafamilial variation is relatively rare 37. Interestingly, members of the same family in our study carried the same variant, but there was significant clinical heterogeneity between them. All the cases in this study had delayed puberty and abnormal teeth, and the first mental symptoms were prominent, with cognitive impairment. Motor function was also affected in the older brothers of the proband. Among them, the proband was characterized by cognitive dysfunction and endocrine abnormality, puberty development was delayed, and the development of breasts and pubes stagnated at Tanner Stage II. After receiving long-term hormone replacement therapy, repeated monitoring of E2, FSH, LH, and P4 remained at low levels. Combined with the results of the GnRH stimulation test and craniocerebral MRI, the gonadotropin deficiency of the proband was considered to be at the pituitary level. Craniocerebral MRI of the proband showed brain atrophy, atrophic degeneration of the white matter of the brain, and atrophy and thinning of the corpus callosum, but no obvious symptoms of ataxia or motor impairment. At the onset of POLR3-HLD, it may only show cognitive dysfunction, but motor symptoms do not exist35. Unlike the proband, her three old brothers have varying degrees of ataxia, dystonia, or dysarthria in addition to cognitive impairment, and have a more rapid progression of impaired motor function. In addition, the proband had microalbuminuria and her father had the phenotype of proteinuria, nephritis, and uremia, while the other members of the family did not have renal complications. POLR3A mutations are associated with WRS, and a case of fatal hyperkalemic renal failure in a three-day-old male infant with features of WRS has been reported, but whether WRS is associated with structural renal anomalies requires more evidence to confirm44. Regrettably, we currently also have no evidence to suggest a correlation between the renal phenotype of this family and the POLR3A c.2300G > T mutant.
As the diagnosis of POLR3-HLD is a combination of typical clinical presentation, brain MRI features, and pathogenic gene testing, the complexity of the clinical phenotype may pose a challenge for early diagnosis45. Dental developmental abnormalities and hypogonadotropic hypogonadism are not prevalent in POLR3-HLD, and in the absence of dental or endocrine abnormalities, brain MRI may better assist in the diagnosis of the disease37,38. When patients have corresponding symptoms or signs, and there are diffuse hypomyelination and cerebellar atrophy on craniocerebral imaging, clinicians should highly doubt the possibility of POLR3-HLD and consider the sequencing of POLR3A and POLR3B genes in time.
In conclusion, HLD-7 is a hereditary hypomyelinating leukodystrophy and its clinical features may differ greatly. As the disease can involve multiple systems, its diagnosis and management need multidisciplinary comprehensive treatment. The intrafamilial variation in this study not only increased the research value of the family to some extent, but also enriched the clinical phenotypic spectrum of POLR3-HLD disease. Although mutants causing POLR3-HLD may affect the normal assembly or biogenesis of Pol III, the functional mechanisms of how these mutations lead to myelin formation deficiency remains to be investigated. In this study, we found that wild-type POLR3A protein overexpression significantly enhanced Pol III transcription of 5S rRNA, tRNA Leu-CAA; however, the increased overexpression of the p.Cys767Phe mutant POLR3A protein compared with the wild-type protein overexpression did not cause the expected further enhancement of Pol III function, but rather frustrated Pol III transcriptional function, suggesting an abnormal function of the mutant POLR3A protein. This study initially confirmed the pathogenicity of the POLR3A p.Cys767Phe mutation, which will provide a reference for further studies on the pathogenic mechanism of POLR3-HLD in the future.
Data availability
The datasets generated and/or analysed during the current study are available in the ClinVAR repository, and the accession number is SCV003928000 (https://www.ncbi.nlm.nih.gov/clinvar/variation/1473885/).
References
Thiffault, I. et al. Recessive mutations in POLR1C cause a leukodystrophy by impairing biogenesis of RNA polymerase III. Nat. Commun. 6, 7623. https://doi.org/10.1038/ncomms8623 (2015).
Schiffmann, R. & van der Knaap, M. S. Invited article: An MRI-based approach to the diagnosis of white matter disorders. Neurology 72, 750–759. https://doi.org/10.1212/01.wnl.0000343049.00540.c8 (2009).
Al Yazidi, G. et al. Dystonia in RNA polymerase III-related leukodystrophy. Mov. Disord. Clin. Pract. 6, 155–159. https://doi.org/10.1002/mdc3.12715 (2019).
Di Donato, I. et al. POLR3A variants in hereditary spastic paraparesis and ataxia: Clinical, genetic, and neuroradiological findings in a cohort of Italian patients. Neurol. Sci. 43, 1071–1077. https://doi.org/10.1007/s10072-021-05462-1 (2022).
Perrier, S., Michell-Robinson, M. A. & Bernard, G. POLR3-related leukodystrophy: Exploring potential therapeutic approaches. Front. Cell. Neurosci. 14, 631802. https://doi.org/10.3389/fncel.2020.631802 (2020).
Dieci, G., Fiorino, G., Castelnuovo, M., Teichmann, M. & Pagano, A. The expanding RNA polymerase III transcriptome. Trends Genet. 23, 614–622. https://doi.org/10.1016/j.tig.2007.09.001 (2007).
Thomas, A. & Thomas, A. K. POLR3-related Leukodystrophy. J. Clin. Imaging Sci. 9, 45. https://doi.org/10.25259/JCIS_116_2019 (2019).
Sosinska-Zawierucha, P., Zawierucha, P., Breborowicz, A. & Barciszewski, J. Prediction of secondary and tertiary structures of human BC200 RNA (BCYRN1) based on experimental and bioinformatic cross-validation. Biochem. J. 475, 2727–2748. https://doi.org/10.1042/BCJ20180239 (2018).
Muslimov, I. A. et al. Autoimmune RNA dysregulation and seizures: Therapeutic prospects in neuropsychiatric lupus. Life Sci. Alliance 5, e202201496. https://doi.org/10.26508/lsa.202201496 (2022).
Borck, G. et al. BRF1 mutations alter RNA polymerase III-dependent transcription and cause neurodevelopmental anomalies. Genome Res. 25, 155–166. https://doi.org/10.1101/gr.176925.114 (2015).
Mendes, M. I. et al. Bi-allelic mutations in EPRS, encoding the glutamyl-prolyl-aminoacyl-tRNA synthetase, cause a hypomyelinating leukodystrophy. Am. J. Hum. Genet. 102, 676–684. https://doi.org/10.1016/j.ajhg.2018.02.011 (2018).
Nakayama, T. et al. Deficient activity of alanyl-tRNA synthetase underlies an autosomal recessive syndrome of progressive microcephaly, hypomyelination, and epileptic encephalopathy. Hum. Mutat. 38, 1348–1354. https://doi.org/10.1002/humu.23250 (2017).
Schaffer, A. E. et al. CLP1 founder mutation links tRNA splicing and maturation to cerebellar development and neurodegeneration. Cell 157, 651–663. https://doi.org/10.1016/j.cell.2014.03.049 (2014).
Musumeci, A. et al. Identification of a novel missense mutation of POLR3A gene in a cohort of sicilian patients with leukodystrophy. Biomedicines 10, 2276. https://doi.org/10.3390/biomedicines10092276 (2022).
Bernard, G. et al. Mutations of POLR3A encoding a catalytic subunit of RNA polymerase Pol III cause a recessive hypomyelinating leukodystrophy. Am. J. Hum. Genet. 89, 415–423. https://doi.org/10.1016/j.ajhg.2011.07.014 (2011).
Zanette, V. et al. Neurodevelopmental regression, severe generalized dystonia, and metabolic acidosis caused by POLR3A mutations. Neurol. Genet. 6, e521. https://doi.org/10.1212/NXG.0000000000000521 (2020).
Tiku, V. & Antebi, A. Nucleolar function in lifespan regulation. Trends Cell Biol. 28, 662–672. https://doi.org/10.1016/j.tcb.2018.03.007 (2018).
Baez-Becerra, C. T. et al. Nucleolar disruption, activation of P53 and premature senescence in POLR3A-mutated Wiedemann-Rautenstrauch syndrome fibroblasts. Mech. Ageing Dev. 192, 111360. https://doi.org/10.1016/j.mad.2020.111360 (2020).
Gutierrez, M. et al. Large exonic deletions in POLR3B gene cause POLR3-related leukodystrophy. Orphanet J. Rare Dis. 10, 69. https://doi.org/10.1186/s13023-015-0279-9 (2015).
Choquet, K. et al. Leukodystrophy-associated POLR3A mutations down-regulate the RNA polymerase III transcript and important regulatory RNA BC200. J. Biol. Chem. 294, 7445–7459. https://doi.org/10.1074/jbc.RA118.006271 (2019).
Choquet, K. et al. Absence of neurological abnormalities in mice homozygous for the Polr3a G672E hypomyelinating leukodystrophy mutation. Mol. Brain 10, 13. https://doi.org/10.1186/s13041-017-0294-y (2017).
Dumay-Odelot, H., Durrieu-Gaillard, S., Da Silva, D., Roeder, R. G. & Teichmann, M. Cell growth- and differentiation-dependent regulation of RNA polymerase III transcription. Cell Cycle 9, 3687–3699. https://doi.org/10.4161/cc.9.18.13203 (2010).
Lata, E. et al. RNA polymerase III subunit mutations in genetic diseases. Front. Mol. Biosci. 8, 696438. https://doi.org/10.3389/fmolb.2021.696438 (2021).
Elbaz, B. & Popko, B. Molecular control of oligodendrocyte development. Trends Neurosci. 42, 263–277. https://doi.org/10.1016/j.tins.2019.01.002 (2019).
Huang, S. et al. Ribosome engineering reveals the importance of 5S rRNA autonomy for ribosome assembly. Nat. Commun. 11, 2900. https://doi.org/10.1038/s41467-020-16694-8 (2020).
Dorboz, I. et al. Mutation in POLR3K causes hypomyelinating leukodystrophy and abnormal ribosomal RNA regulation. Neurol. Genet. 4, e289. https://doi.org/10.1212/NXG.0000000000000289 (2018).
Roder, K., Stirnemann, G., Dock-Bregeon, A. C., Wales, D. J. & Pasquali, S. Structural transitions in the RNA 7SK 5’ hairpin and their effect on HEXIM binding. Nucleic Acids Res. 48, 373–389. https://doi.org/10.1093/nar/gkz1071 (2020).
Briese, M. & Sendtner, M. Keeping the balance: The noncoding RNA 7SK as a master regulator for neuron development and function. Bioessays 43, e2100092. https://doi.org/10.1002/bies.202100092 (2021).
Azmanov, D. N. et al. Transcriptome-wide effects of a POLR3A gene mutation in patients with an unusual phenotype of striatal involvement. Hum. Mol. Genet. 25, 4302–4314. https://doi.org/10.1093/hmg/ddw263 (2016).
Yeganeh, M. & Hernandez, N. RNA polymerase III transcription as a disease factor. Genes Dev. 34, 865–882. https://doi.org/10.1101/gad.333989.119 (2020).
Booy, E. P., McRae, E. K., Koul, A., Lin, F. & McKenna, S. A. The long non-coding RNA BC200 (BCYRN1) is critical for cancer cell survival and proliferation. Mol. Cancer 16, 109. https://doi.org/10.1186/s12943-017-0679-7 (2017).
McLaurin, J., Trudel, G. C., Shaw, I. T., Antel, J. P. & Cashman, N. R. A human glial hybrid cell line differentially expressing genes subserving oligodendrocyte and astrocyte phenotype. J. Neurobiol. 26, 283–293. https://doi.org/10.1002/neu.480260212 (1995).
Boggs, J. M. Myelin basic protein: A multifunctional protein. Cell. Mol. Life Sci. 63, 1945–1961. https://doi.org/10.1007/s00018-006-6094-7 (2006).
Voccoli, V., Tonazzini, I., Signore, G., Caleo, M. & Cecchini, M. Role of extracellular calcium and mitochondrial oxygen species in psychosine-induced oligodendrocyte cell death. Cell Death Dis. 5, e1529. https://doi.org/10.1038/cddis.2014.483 (2014).
Campopiano, R. et al. A novel POLR3A genotype leads to leukodystrophy type-7 in two siblings with unusually late age of onset. BMC Neurol. 20, 258. https://doi.org/10.1186/s12883-020-01835-9 (2020).
Tewari, V. V. et al. A novel homozygous mutation in POLR3A gene causing 4H syndrome: A case report. BMC Pediatr. 18, 126. https://doi.org/10.1186/s12887-018-1108-9 (2018).
Wolf, N. I. et al. Clinical spectrum of 4H leukodystrophy caused by POLR3A and POLR3B mutations. Neurology 83, 1898–1905. https://doi.org/10.1212/WNL.0000000000001002 (2014).
Steenweg, M. E. et al. Magnetic resonance imaging pattern recognition in hypomyelinating disorders. Brain 133, 2971–2982. https://doi.org/10.1093/brain/awq257 (2010).
Vrij-van den Bos, S. et al. 4H leukodystrophy: A brain magnetic resonance imaging scoring system. Neuropediatrics 48, 152–160. https://doi.org/10.1055/s-0037-1599141 (2017).
Wu, S. et al. Novel mutations of the POLR3A gene caused POLR3-related leukodystrophy in a Chinese family: A case report. BMC Pediatr. 19, 289. https://doi.org/10.1186/s12887-019-1656-7 (2019).
La Piana, R. et al. Diffuse hypomyelination is not obligate for POLR3-related disorders. Neurology 86, 1622–1626. https://doi.org/10.1212/WNL.0000000000002612 (2016).
Harting, I. et al. POLR3A variants with striatal involvement and extrapyramidal movement disorder. Neurogenetics 21, 121–133. https://doi.org/10.1007/s10048-019-00602-4 (2020).
La Piana, R. et al. Brain magnetic resonance imaging (MRI) pattern recognition in Pol III-related leukodystrophies. J. Child Neurol. 29, 214–220. https://doi.org/10.1177/0883073813503902 (2014).
Ghamry, M. A., Salah, R., Galal, E. I., Henin, S. & Dobs, M. A case of Wiedemann-Rautenstrauch syndrome with fatal hyperkalemic renal faliure. Cureus 14, e29320. https://doi.org/10.7759/cureus.29320 (2022).
Billington, E., Bernard, G., Gibson, W. & Corenblum, B. Endocrine aspects of 4H leukodystrophy: A case report and review of the literature. Case Rep. Endocrinol. 2015, 314594. https://doi.org/10.1155/2015/314594 (2015).
Funding
This work was supported by Fujian Province Medical Innovation Foundation (2021CXB001, 2022CXB002, 2022CXA001), Fujian Province Natural Science Fund Project (2022J01996, 2021J02053, 2021J01704), Joint Funds for the innovation of science and Technology in Fujian province (2023Y9284), the Special Research Foundation of Fujian Provincial Department of Finance , China (2020-822, 2021-848, 2022-840), and National famous and old Chinese medicine experts (Zhang Xuemei, Yan Xiaohua) inheritance studio construction project.
Author information
Authors and Affiliations
Contributions
Z.Z., Y.F.L., F.L., and J.W.L. conceived and designed the study. D.D.R., M.W., J.H.Z., Y.W.Z., and Y.F.L. collected the data. Y.P.Z., B.L., S.J.L., and Q.C. conducted the data analysis and interpretation. D.D.R., X.L.R., and R.L.W. wrote the manuscript. X.L.R., Q.C., X.F.L., and J.W.L. made critical revisions to the important contents of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ruan, Dd., Ruan, Xl., Wang, R. et al. Clinical phenotype and genetic function analysis of a family with hypomyelinating leukodystrophy-7 caused by POLR3A mutation. Sci Rep 14, 7638 (2024). https://doi.org/10.1038/s41598-024-58452-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-58452-6
Keywords
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
