
Abstract
Leukodystrophies are a heterogenous group of genetic disorders, characterised by abnormal development of cerebral white matter. Pelizaeus-Merzbacher disease is caused by mutations in PLP1, encoding major myelin-resident protein required for myelin sheath assembly. We report a missense variant p.(Ala109Asp) in MAL as causative for a rare, hypomyelinating leukodystrophy similar to Pelizaeus-Merzbacher disease. MAL encodes a membrane proteolipid that directly interacts with PLP1, ensuring correct distribution during myelin assembly. In contrast to wild-type MAL, mutant MAL was retained in the endoplasmic reticulum but was released following treatment with 4-phenylbutyrate. Proximity-dependent identification of wild-type MAL interactants implicated post-Golgi vesicle-mediated protein transport and protein localisation to membranes, whereas mutant MAL interactants suggested unfolded protein responses. Our results suggest that mislocalisation of MAL affects PLP1 distribution, consistent with known pathomechanisms for hypomyelinating leukodystrophies.
Similar content being viewed by others
Introduction
Genetic forms of leukodystrophy are a heterogenous group of congenital hypomyelinating or demyelinating disorders, resulting from deficient or abnormal myelin deposition within the CNS. Leukodystrophies have varied clinical and radiological phenotypes, and the largest group are hypomyelinating leukodystrophies that have significant and permanent deficit of myelin. PLP1 mutations are the most common, causative for Pelizaeus-Merzbacher disease [1]. PLP1 encodes proteolipid protein 1, the major myelin-resident protein required for myelin sheath assembly in nerve fibres [2]. Correctly-timed trafficking of PLP1 is fundamental to myelin biogenesis during development [3].
MAL (myelin and lymphocyte protein) encodes a membrane proteolipid with four transmembrane domains mainly localised in compact myelin that is highly expressed in pre-mature Schwann cells, oligodendrocytes and mature Schwann cells [4]. MAL directly interacts with PLP1, redirecting PLP1 transport towards myelin membranes [2]. MAL also mediates vesicular trafficking including direct apical transport from the Golgi apparatus (essential for myelination by oligodendrocytes) and transcytosis to basolateral membranes [5].
Here, we report the identification and characterisation of a missense variant in MAL as causative for a rare, hypomyelinating leukodystrophy. As previously observed for PLP1 mutations, mutated MAL protein was retained in the endoplasmic reticulum (ER) [6, 7] and was unable to bind PLP1. Our results suggest that mislocalisation of MAL affects the distribution of PLP1, consistent with known pathomechanisms for Pelizaeus-Merzbacher disease.
Materials and methods
Patients
Family members were recruited with informed consent and under ethical approval from South Yorkshire Research Ethics Committee (REC ref. no. 11/H1310/1). Genomic DNA was obtained from blood samples as described previously [8].
Whole exome sequencing and bioinformatics analysis
Whole exome sequencing (WES) was performed using genomic DNA from two affected and one unaffected sibling (indicted by *, Fig. 1A) in the family. DNA was processed using the Agilent ‘SureSelect QXT’ Target Enrichment kit (Agilent Technologies). DNA libraries were sequenced on an Illumina HiSeq 3000 using a 150 bp paired-end protocol. Bioinformatics analysis was done using standard pipelines (Supplementary Methods A).

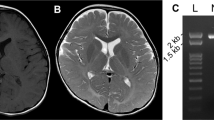
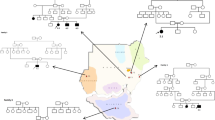
A Pedigree of the family under study, summarising four generations and the consanguineous union of the parents of the two affected individuals (IV3 and IV5). Individuals recruited to the study are indicated by Roman numerals, and individuals whose DNA samples have been used for WES are marked with a star (*). B–G MR images of individual IV3 (B–D) aged 14 years, and his brother IV5 (E–G) aged 6 years. T2 axial images (B, C & E, F) demonstrating lack of hemispheric myelin visible through T2 hyperintensity (brightness indicated by white arrows). Prominence of cerebellar folia and vermian hypoplasia (C & F) indicates cerebellar volume loss. T1 sagittal images (D & G) demonstrate a thin corpus callosum (D, indicated by white arrowhead) and mild upper vermian volume loss. H Ideogram illustrating the shared homozygous regions of the two affected individuals in red, and a table summarising the significant homozygous regions in chromosome order. The largest homozygous region on chromosome 2 that contains MAL is marked by the red asterisk (*). I Segregation analysis of the MAL c.326 C > A p.(Ala109Asp) variant (indicated by red asterisk, *) within the family confirmed by Sanger sequencing. het heterozygous, ho homozygous.
Molecular biology and other methods
PCR, Sanger sequencing, cDNA insert cloning and site-directed mutagenesis are summarised in Supplementary Methods B–C. Cell culture, transfection, western blotting and immunoprecipitation methodologies were performed as described previously [8], summarised in Supplementary Methods D–F. BioID2 and mass spectroscopy analysis is outlined in Supplementary Methods G. Confocal imaging and live cell imaging are detailed in Supplementary Methods H. Statistical analysis is summarised in Supplementary Methods I.
Results
Clinical ascertainment
Two affected male siblings presented with developmental delay and nystagmus in infancy, followed by significant learning disabilities and progressive motor deterioration within the first decade. The older sibling also had epilepsy. Both patients were described to be increasingly unsteady with falls. Examination of the younger sibling aged 8 demonstrated increased lower limb tone, brisk deep tendon reflexes and new onset ataxia. He had intention tremor and mild dysmetria. MR imaging demonstrated cerebellar volume loss with patchy dysmyelination in subcortical areas in T2 sequences (Fig. 1A–G) that is different from Pelizeaus-Merzbacher patients who usually have a diffuse hypomyelination, mainly in periventricular regions and semi-ovale centres. Sequencing of PLP1 did not reveal pathogenic mutations and an MLPA assay excluded PLP1 duplication. No further mutations were identified in a diagnostic panel for white matter-associated genes (Supplemental Data A). The parents of the affected siblings were first cousins of Pakistani origin (Fig. 1A).
Whole exome sequencing and in silico modelling
Whole exome sequencing was performed on the two affected boys and an unaffected sibling. Variants incompatible with autosomal recessive or X-linked inheritance were filtered out. Autozygosity mapping allowed prioritisation of variants within regions identical-by-descent (Fig. 1H), and three variants were identified that passed filtering criteria (Supplementary Data B). One variant did not segregate in the family and a second was in a gene, ZNHIT1, that was an unlikely functional candidate (Supplementary Fig. 1). A homozygous variant in MAL, p.(Ala109Asp), segregated in the family (Fig. 1I) was predicted to be pathogenic by Condel, Polyphen2 and SIFT, had a CADD score of 34, and was absent from dbSNP151 and gnomAD (v2.1.1). In silico modelling MAL p.(Ala109Asp) predicted alpha-helical secondary structure was lost (HeliQuest) preventing insertion of the third transmembrane domain into a membrane (Transmembrane Helix Prediction TMHMM Server v.2.0; Fig. 2A).

A Computational prediction of the effect of the p.(Ala109Asp) variant on the transmembrane helix organisation of the MAL protein using the TMHMM Transmembrane Helix prediction tool (left). Top image indicates that each of the transmembrane helices (red) in the wildtype MAL protein have the predicted organisation of a tetraspanin-like protein, but the third transmembrane domain in p.(Ala109Asp) mutant MAL is predicted to no longer span the membrane as a result of the hydrophilic amino acid substitution. HeliQuest validated this change (right) showing that the hydrophobic phase of the wildtype helix (LFYAAAM; residues coloured yellow) is compromised by the mutation (D; red). B Live cell imaging of MAL wildtype and MAL p.(Ala109Asp) mutant C-terminal eYFP-tagged proteins, visualised using a Nikon BioStation system for a total of 24 h. The wildtype MAL protein localised at the cell membrane and some perinuclear regions, whereas mutant MAL protein formed prominent aggregates in perinuclear regions of the ER (arrow). Scale bar = 10 μm. C Immunofluorescence staining and confocal microscopy imaging of wildtype and mutant MAL eYFP-tagged protein overexpression in MDCK cells, following 4-phenylbutyrate (4-PBA) treatment, calreticulin (red) and DAPI (blue). Co-localisation of mutant MAL-eYFP and calreticulin (detail visible in magnified insets; indicated by white boxes) in ER aggregate (arrows) is reduced following 4-PBA treatment. IF experiments were performed in three independent biological replicates, each of three technical replicates each, with analysis of a total of 120 cells. Scale bar = 20 μm. D Top graph: co-localisation analysis of MAL protein and the ER marker calreticulin, using Fiji to determine Pearson R2 “Rcoloc” correlation values for each transfected cell. There is a significant increase in ER colocalization for mutant MAL protein compared to the wildtype. Analysis included a total of 100 cells, from three independent biological replicates. Statistical analysis of a pairwise comparison was performed using a two-tailed Student t test (****p < 0.0001). Error bars indicate s.e.m. Bottom graph: analysis of MAL protein and ER colocalization, for control cells or following 4-PBA treatment. Rcoloc values for each cell analysed indicate a significant difference in ER colocalization for the treated cells expressing mutant MAL compared to the untreated control cells expressing mutant MAL. Statistical analysis of pair-wise comparisons was performed by a two-tailed Student t test (ns not significant; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001). Error bars indicate s.e.m.
MAL p.(Ala109Asp) forms aggregates in the endoplasmic reticulum that are decreased by the chemical chaperone 4-phenylbutyrate
Wild-type MAL-eYFP fusion protein localised to perinuclear and basolateral lineate localisations at the plasma membrane in polarised MDCK cells (Fig. 2B), whereas mutant MAL protein co-localised in aggregates containing calreticulin, a marker of the ER (Fig. 2C–D). ER protein aggregates form when newly-synthesised proteins fail to fold correctly, leading to ER stress and activation of unfolded protein responses (UPR) [9, 10]. Treatment with 4-phenylbutyrate, a chemical chaperone, significantly decreased co-localisation of mutant MAL p.(Ala109Asp) protein with calreticulin (Fig. 2C–D) suggesting that it corrected protein folding.
Missense variant p.(Ala109Asp) abrogates the interaction with PLP1 and correct distribution to basolateral membranes
In oligodendrocytes, MAL acts as a regulator of PLP1 transcytosis during myelin formation [2, 3]. Quantification of immunoprecipitation assays following co-expression of MAL-V5 and PLP1-GFP-tagged proteins revealed a 56% loss of interaction between mutant MAL and PLP1 (Fig. 3A–B). Next, we used the EZ-Link-Sulfo-NHS-Biotin assay to biotinylate lysine residues in apical cell surface proteins (Fig. 3C). Apically mislocalized PLP1 was detected in cells expressing mutant MAL whereas wild-type protein was hardly detectable, suggesting that correct redirection of PLP1 from apical to basolateral membranes only occurs in cells expressing wildtype MAL.

A Western blot analysis of immunoprecipitations to assess interactions between PLP1-GFP protein and either wildtype or mutant MAL C-terminal V5-tagged protein. Pull-down was performed with anti-GFP beads and westerns blots resolved with anti-V5 antibody. The arrow indicates MAL protein (expected size 16 kDa). B Quantification of wildtype or mutant MAL and PLP1 interactions from three independent biological replicates. Statistical analysis of pair-wise comparisons was performed by a two-tailed Student t test (**p < 0.01). Error bars indicate s.e.m. C Schematic of the EZ-Link-Sulfo-NHS-Biotin biotinylation assay, in order to assess the localisation of PLP1 for cells expressing either wildtype or mutant MAL. On the left, PLP1 correctly localises to the basolateral membrane in cells expressing wildtype MAL but remains in the apical membrane in the mutant MAL model. D Western blot results from the biotinylation assay showing a reduction in PLP1 levels from the apical membrane for cells expressing wildtype MAL compared to mutant MAL model. Total input PLP1 levels are determined by anti-PLP1 western blotting of whole cell extracts (WCE) and β-actin is a loading control. E Quantification of the western blot results using Image Lab. Anti-PLP1 band intensity from the pull-down was normalised to anti-PLP1 bands in WCE, corrected for overall loading using β-actin levels. Data presented is for two independent biological replicates. F Proposed mechanism of the role of MAL as a regulator of intracellular PLP1 trafficking. Nascent PLP1 is trafficked from the ER to the apical cell membrane (arrow 1). MAL and PLP1 interact at the apical membrane of myelinating oligodentrocytes (arrow 2), where MAL redirects PLP1 localisation to the basolateral membrane (arrow 3) during myelin formation.
Proximity-dependent interactants of wild-type and mutant MAL
Expression of wild-type and mutant MAL-BirA2 (Supplementary Fig. 2) for ‘BioID’ proximity-dependent biotinylation identified candidate interacting proteins required for PLP1 transcytosis. Lysates were pulled-down with streptactin beads, trypsinised and then analysed using mass spectroscopy (Supplementary Data C). MAL and PLP1 proteins had limited trypsin cleavage sites, and their presence in the pull-downs was confirmed using western blotting (Supplementary Fig. 3). Analysis by STRING [11] identified interactants of wild-type MAL associated with post-Golgi vesicle-mediated protein transport, macromolecule localisation and protein localisation to membranes (Supplementary Fig. 4A). Pathway enrichment analysis using Database for Annotation, Visualisation and Integrated Discovery (DAVID v6.8) [12] identified plasma membrane components, intracellular vesicle-mediated transport, lipoprotein and membrane raft pathways (Supplementary Fig. 5A), consistent with the role of wild-type MAL as a lipoprotein involved in regulating PLP1 during trafficking from ER to membrane. Interactants of mutant MAL (Supplementary Fig. 4B) were enriched for pathways that included phagosome acidification and macroautophagy (Supplementary Fig. 5B), suggesting these proteins mediated ER stress or UPR as a result of MAL aggregates.
Discussion
We present a missense variant in MAL as the likely cause of hypomyelinating leukodystrophy, resulting in ER mislocalization of mutant MAL leading to possible defects in PLP1 trafficking. One function of the ER is to assess the quality of newly-synthesised proteins, with misfolded proteins tending to form aggregates leading to ER stress [9]. ER stress then activates the UPR, in order to mitigate the stress by degrading the ER aggregates, diminishing protein translation and increasing the expression of ER chaperones [10, 13]. If the UPR fails, then cumulative ER stress will eventually lead to apoptosis [14]. The formation of ER aggregates caused by point mutations is a common disease mechanism for proteinopathies [15, 16], and is a possible pathomechanism for the MAL missense mutation p.(Ala109Asp). Our data suggests that p.(Ala109Asp) severely affects protein folding of MAL, leading to mislocalization in the ER. This was partly resolved by treatment with 4-phenylbutyrate, likely mediating correct folding of mutant MAL [17].
‘BioID’ proximity-dependent biotinylation identified potential protein interactions that provide insights into MAL function and how the p.(Ala109Asp) mutation affects these interactions. Interactions with wild-type MAL were grouped into cellular processes involving mainly vesicular transport. An example is VAPB, a protein mediating vesicle trafficking that is implicated in amyotrophic lateral sclerosis [18] and spinal muscular atrophy [19]. Another interactant was CKAP5, a cytoskeleton-associated protein, involved in the translation of myelin basic protein (MBP) consistent with a central role for MAL in myelin formation during neurodevelopment.
MAL acts as a regulator of PLP1 trafficking, redirecting PLP1 transport towards basolateral membranes during myelin formation [2]. In oligodendrocytes, MAL is not involved in PLP1 transport from the Golgi to the apical membrane, but it appears to redirect PLP1 trafficking from the apical membrane to the basolateral membrane where myelin begins to form [3]. Our data suggests that mutant p.(Ala109Asp) MAL, mislocalized in the ER, does not reach apical membranes where MAL and PLP1 interact. Loss of this interaction likely affects PLP1 transcytosis and subsequent myelin formation (Fig. 3C, E), thereby suggesting that the p.(Ala109Asp) missense variant is likely pathogenic on the basis of altered protein function.
In conclusion, this study describes the identification of a missense mutation in MAL that causes a neurodevelopmental condition characterised by hypomyelination and cerebellar atrophy. Our work supports a disease mechanism for leukodystrophies by which mislocalisation of MAL affects the distribution of PLP1 [20], resulting in a hypomyelination disorder similar to Pelizaeus-Merzbacher disease. This is consistent with the suggested pathomechanisms for hypomyelination that include defects in membrane integration or membrane interactions between proteins [20], for example transmembrane protein 106B (TMEM106B) that appears to mediate PLP1 trafficking.
Data availability
The datasets generated and analysed during the current study are available from the corresponding author on reasonable request.
References
Osorio MJ, Goldman SA. Neurogenetics of Pelizaeus-Merzbacher disease. Handb Clin Neurol. 2018;148:701–22.
Bijlard M, de Jonge JC, Klunder B, Nomden A, Hoekstra D, Baron W. MAL is a regulator of the recruitment of myelin protein PLP to membrane microdomains. Plos ONE. 2016;11:e0155317.
Baron W, Ozgen H, Klunder B, de Jonge JC, Nomden A, Plat A, et al. The major myelin-resident protein PLP is transported to myelin membranes via a transcytotic mechanism: involvement of sulfatide. Mol Cell Biol. 2015;35:288–302.
Frank M. MAL, a proteolipid in glycosphingolipid enriched domains: functional implications in myelin and beyond. Prog Neurobiol. 2000;60:531–44.
Puertollano R, Alonso MA. MAL, an integral element of the apical sorting machinery, is an itinerant protein that cycles between the trans-Golgi network and the plasma membrane. Mol Biol Cell. 1999;10:3435–47.
Inoue K. Cellular pathology of pelizaeus-merzbacher disease involving chaperones associated with endoplasmic reticulum stress. Front Mol Biosci. 2017;4:7.
Nevin ZS, Factor DC, Karl RT, Douvaras P, Laukka J, Windrem MS, et al. Modeling the mutational and phenotypic landscapes of pelizaeus-merzbacher disease with human iPSC-derived oligodendrocytes. Am J Hum Genet. 2017;100:617–34.
Elpidorou MBS, Poulter JA, Hartill V, Hobson E, Sheridan E, Johnson CA. Novel loss-of-function mutation in HERC2 is associated with severe developmental delay and paediatric lethality. J Med Genet. 2021;58:334–41.
Ihara S, Nakayama S, Murakami Y, Suzuki E, Asakawa M, Kinoshita T, et al. PIGN prevents protein aggregation in the endoplasmic reticulum independently of its function in the GPI synthesis. J Cell Sci. 2017;130:602–13.
Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science.2011;334:1081–6.
Snel B, Lehmann G, Bork P, Huynen MA. STRING: a web-server to retrieve and display the repeatedly occurring neighbourhood of a gene. Nucleic Acids Res. 2000;28:3442–4.
Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57.
Schröder M, Kaufman RJ. The mammalian unfolded protein response. Annu Rev Biochem. 2005;74:739–89.
Tabas I, Ron D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat Cell Biol. 2011;13:184–90.
Brookes SJ, Barron MJ, Smith CEL, Poulter JA, Mighell AJ, Inglehearn CF, et al. Amelogenesis imperfecta caused by N-terminal enamelin point mutations in mice and men is driven by endoplasmic reticulum stress. Hum Mol Genet. 2017;26:1863–76.
Xiang CC, Wang YJ, Zhang H, Han F. The role of endoplasmic reticulum stress in neurodegenerative disease. Apoptosis.2017;22:1–26.
Mimori S, Okuma Y, Kaneko M, Kawada K, Hosoi T, Ozawa K, et al. Protective effects of 4-phenylbutyrate derivatives on the neuronal cell death and endoplasmic reticulum stress. Biol Pharm Bull. 2012;35:84–90.
Kanekura K, Nishimoto I, Aiso S, Matsuoka M. Characterization of amyotrophic lateral sclerosis-linked P56S mutation of vesicle-associated membrane protein-associated protein B (VAPB/ALS8). J Biol Chem. 2006;281:30223–33.
Nishimura AL, Mitne-Neto M, Silva HCA, Richieri-Costa A, Middleton S, Cascio D, et al. A mutation in the vesicle-trafficking protein VAPB causes late-onset spinal muscular atrophy and amyotrophic lateral sclerosis. Am J Hum Genet. 2004;75:822–31.
Wolf NI, Ffrench-Constant C, van der Knaap MS. Hypomyelinating leukodystrophies - unravelling myelin biology. Nat Rev Neurol. 2021;17:88–103.
Acknowledgements
The authors would like to thank the family and the patients for participating in this study.
Funding
This study was funded by a Sir Jules Thorn Award for Biomedical Research (JTA/09 to EGS and CAJ), Medical Research Council (MR/M000532/1 to CAJ) and the University of Leeds. WB acknowledges funding from Stichting MS Research. MU was supported by the Tistou & Charlotte Kerstan Stiftung. JAP was supported by a UKRI Future Leaders Fellowship (MR/T02044X/1).
Author information
Authors and Affiliations
Contributions
Conceptualisation: JAP, EGS, CAJ, GW; Data curation: MA, KS, KJ, KB, MU, LG; Formal analysis: KB, MU, LG, JHL, EGS, CAJ; Funding acquisition: JAP, JHL, EGS, CAJ; Investigation: MA, KS, KJ, KB, CAJ; Methodology: MA, KS, WB, KB, LG, JHL, EGS, CAJ; Project administration: MA, JAP, EGS, CAJ; Resources: KS, WB, KJ, KB, MU; Supervision: JAP, EGS, CAJ; Writing – original draft: MA, JAP, EGS, CAJ; Writing – review and editing: all authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
Family members were recruited with informed consent and under ethical approval from South Yorkshire Research Ethics Committee (REC ref. no. 11/H1310/1).
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Elpidorou, M., Poulter, J.A., Szymanska, K. et al. Missense mutation of MAL causes a rare leukodystrophy similar to Pelizaeus-Merzbacher disease. Eur J Hum Genet 30, 860–864 (2022). https://doi.org/10.1038/s41431-022-01050-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-022-01050-9
This article is cited by
-
Clinical genomics testing: mainstreaming and globalising
European Journal of Human Genetics (2022)
