
Abstract
Endometrial epithelial cells carry distinct cancer-associated alterations that may be more susceptible to endometriosis. Mouse models have shown that overexpression of SIRT1 associated with oncogene activation contributes to the pathogenesis of endometriosis, but the underlying reason remains elusive. Here, we used integrated systems biology analysis and found that enrichment of endometrial stromal fibroblasts in endometriosis and their cellular abundance correlated negatively with epithelial cells in clinical specimens. Furthermore, endometrial epithelial cells were characterized by significant overexpression of SIRT1, which is involved in triggering the EMT switch by escaping damage or oncogene-induced induced senescence in clinical specimens and in vitro human cell line models. This observation supports that genetic and epigenetic incident favors endometrial epithelia cells escape from senescence and fuel EMT process in endometriosis, what could be overcome by downregulation of SIRT1.
Similar content being viewed by others


Introduction
Endometriosis is characterized by the growth of endometrial glands and stroma-like tissue outside of the uterine cavity. Although controversies remain regarding the genesis of endometriotic lesions1,2, it is most commonly accepted that the endometrium is the main reason and that some cellular or genomic incident must have occurred to cause endometriosis1. At endometriotic sites, the ectopic glands always histologically resemble uterine endometrial glands but show variations in different epithelial-to-stromal cell ratios to the extreme case of stromal endometriosis3.
One of the mechanisms suggested to participate in the onset of endometriosis is the epithelial-mesenchymal transition (EMT)4,5. EMT is a process by which epithelial cells convert to motile mesenchymal cells. This mostly occurs during embryonic development and cancer and has been found in endometrial epithelial cells in vitro and in human endometrial tissues6. Once the ectopic endometriotic lesion is established, multiple mechanisms involved with EMT are activated, resembling cancer-like progression and invasive growth7. It is also well known that EMT endows epithelial cells with migratory and invasive properties, similar to metastatic carcinoma cells8. Furthermore, the cancer-associated mutations and/or activation are thought to have metastatic potential, are present in endometriosis and appear to be confined to the epithelial but not the stromal compartment9.
The human endometrium is a highly regenerative tissue undergoing growth and shedding throughout the menstrual cycle; both the senescence program and cancer-associated mutations are involved in endometrial regeneration9,10, and when perturbed, they can have detrimental effects during tumour progression and ageing11,12. Meanwhile, abnormal levels of cellular senescence in endometrial epithelial cells may be one of the causes of reproductive diseases, including miscarriage and endometriosis13,14. Moreover, it has been shown that the acquisition of EMT may be accompanied by the bypass of senescence in most cancers15,16,17. Due to the invasive behaviour and ageing phenotypes, we reasoned that escape from senescence of endometrial epithelial cells might contribute to the acquisition of metastatic features in endometriosis.
Sirtuin family proteins are class Ш histone deacetylases (HDACs) and the best known is the well-described role of SIRT1 in cancer and ageing18. Thus, we hypothesized that escape from senescence mediated by SIRT1 is associated with the epithelial to mesenchymal transition in endometriosis.
Results
Bioinformatic analysis showed that EMT and senescence are involved in endometriosis
To gain insights into the pathological changes during endometriosis, we analysed differentially expressed genes (DEGs) in the public dataset GSE7305, GSE11691 GSE5108 and E-MEXP-1251, which contains microarray data of endometriotic and normal endometrial specimens (Fig. 1A and Supplementary Figs. S1–S3). Pathway enrichment analysis identified significant dysregulation of the previously described crosstalk between senescence and EMT pathways19,20, such as “p53”, “MAPK” and “cell cycle” (Fig. 1B). Furthermore, GSEA was performed to explorer differences in endometriosis and and its related signal pathway for senescence and EMT (Fig. 1C). KEGG enrichment analysis showed that endometriosis was associated with the cell cycle (Fig. 1D).The HALLMARK gene sets showed positive enrichment of genes in EMT and positive enrichment of genes in cell cycle arrest, including the cell cycle, G2/M checkpoint and DNA repair (Fig. 1E), which are known to be linked to cellular senescence.

Bioinformatic analysis results support that EMT and senescence are involved in endometriosis. Differentially expressed genes (DEGs) were identified. The red and green indicate unregulated and downregulated DEGs, respectively, in GSE7305 (A). Hallmark pathway enrichment (B) and heatmap of EMT- and senescence-related genes from GSE7305 (C). Endometriosis showing enhanced genes in the “cell cycle” pathway (D). GSEA revealed enrichment of endometriosis genes in “EMT”, “DNA repair” and “G2/M checkpoint” (E).
We then examined the expression levels of EMT- and senescence-related genes in clinical endometriotic specimens and found that both the mRNA and protein levels of EMT-related genes (E-cadherin, vimentin and ZEB2) were abnormally expressed in ectopic endometrial tissues (Fig. 2A, n = 10). In contrast, senescence-related gene levels (p53, p16 and p38 MAPK) were suppressed (Fig. 2B, n = 10).

Relative expression of EMT- and senescence-related genes Expression as shown by qRT-PCR and western blotting, respectively (A,B). Anti-GAPDH antibody was used as loading control. Greyscale values of the bands were determined by Quantity One software. *p < 0.05, **p < 0.01, ***p < 0.001.
Senescence and EMT occur in epithelial cells of of endometriosis
Since the presence of numerous cell types within samples can influence gene signatures, we applied the xCell package to determine the tissue cellular heterogeneity of endometriosis in these four datasets (Fig. 3 and Supplementary Fig. S4). In GSE11691, the most prevalent cells were fibroblasts, stem cells and endothelial cells, and very few epithelial cells were found in endometriosis (Fig. 3A). We then compared the frequency of each cell type between endometriotic and normal endometrium, which indicated that there was a significantly different composition of the cell types between endometriosis and control, and fibroblasts were enriched (the most abundant) cell type in endometriotic endometrium, while epithelial cells were enriched (the most abundant) cell type in control endometrium (Fig. 3B). For external validation, we confirmed a decrease in epithelial cells and an increase in fibroblast abundance in endometriosis compared with control tissues in both GSE7305 and GSE5108, as well as in endometrial tissues (Fig. 3B,C). The number of epithelial cells negatively correlated with the number of fibroblasts in tissues of the control and endometriosis in GSE11691 (Fig. 3D). The full list of scores for all specific cell types is presented in the Supplementary Table S1. Similar results were also noted in other three dataset (Supplementary Fig. S5). Overall, the fact that epithelial cells can transform into stromal cells supports that they may contribute to the high fibroblast abundance and the pathogenesis of endometriosis.

Endometrial stromal fibroblasts were enriched in endometriosis compared to other cell types. Computational biology analysis revealed the total content of different cells in the tissues of the control and endometriosis (GSE11691, n = 10) (A). Endometrial stromal fibroblast and epithelial cell abundance scores in tissues of the control and endometriosis, as calculated by xCell (B). HE staining and vimentin immunohistological staining showing stromal fibroblasts and epithelial cells (C). Abundance score correlation analysis for stromal fibroblasts with epithelial cells in tissues of the control and endometriosis (D). Scale bars = 50 µm in (C). ***p < 0.001.
Although the impact of aging on uterine function is less clear21, it has been demonstrated that endometrial epithelial cells display characteristic features of senescence in cyclic endometrium22, whereas in endometrial cancer, cell escapes from senescence and acquires the aggressive phenotype23. Based on these observations, we evaluated senescence and EMT markers using immuhistochemistry in endometrial specimens. Consistent with the previous studies6, epithelial cells in ectopic lesions showed a decrease of E- cadhein (a hallmark of EMT) and an increase of vimentin (mesenchymal marker). In contrast, the expression of senescence markers including β-gal, p16 and p38 MAPK were decreased in ectopic lesions compared to normal and eutopic endometriotic lesions (Fig. 4A–C).

EMT- and senescence-related protein expression in epithelial cells of endometriosis. Immunostaining for E-cad, vimentin, β-gal, p16 and p38 (A). The intensity of the immunohistological staining was given an IRS score as the mean ± SEM. The results show that E-cad and vimentin expression was stronger, but β-gal, p16 and p38 expression was weaker in epithelial cells in endometriosis tissues than in the controls (B). E-cad and vimentin protein levels were examined by immunofluorescence staining (C). Scale bars = 50 µm in (A,C). ***p < 0.001.
Both the previous literature and human samples highlight the correlation between SIRT1 upregulation and endometriosis
Having demonstrated the decreased cellular senescence and enhanced EMT in endometriosis, we then aimed to investigate the molecular mediators of the process. Although previous literature7,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42 demonstrated that EMT and endometriosis are closely associated with SIRT1 genes, indicating the involvement of SIRT1 in the pathogenesis of endometriosis (Table 1), no study has been performed to explore whether SIRT1 regulates EMT and senescence in endometriosis. We thus examined the effect of SIRT1 on the associated differential gene expression. In dataset GSE7305, the volcano plot heighted that SIRT1, ZEB2, p53 and E-caherin were the significantly differentially expressed between the endometriosis group and the control group (Fig. 5A). SIRT1 and ZEB2 were differentially expressed with significantly higher expression in endometriotic specimens (FC = 1.29, p = 0.005; FC = 0.83, p < 0.001, respectively), while E-cadherin and p53 showed significantly lower expression in endometriotic specimens (FC = 0.13, p = 0.016; FC = 0.39, p < 0.001, respectively) (Fig. 5A). Correlation analysis was used to determine the degree of association. SIRT1 expression was positively correlated with ZEB2 (p = 0.01, R2 = 0.31), while SIRT1 expression was inversely correlated with E-cadherin (p = 0.05, R2 = 0.18), p53 expression (p = 0.008, R2 = 0.33), and p16 expression (p = 0.007, R2 = 0.34) (Fig. 5B), indicating the underlying regulatory mechanism. Similar results were also observed in the GSE11691, GSE5108 and E-MEXP-1251 dataset (Supplementary Fig. S6).

The expression of SIRT1 was unregulated in endometriosis. A volcano plot shows the high expression of SIRT1 and ZEB2 and the low expression of E-cad and p53 in endometriosis (A). Correlation analysis of SIRT1 with ZEB2, E-cad, p53 and p16 in endometriosis (B). Representative images of SIRT1 expression were detected by IHC in control and endometriotic endometria (C). Dot plot of the IRS scores. The differences were analysed by Student’s t-test. ***p < 0.001 (D). qPCR (E) and western blots (F) were used to detect the SIRT1 expression levels in the control and endometriotic endometrium. E-cad E-cadherin, EM endometriosis. Scale bars = 50 µm in (C).
The expression of SIRT1 was investigated in endometriosis and control tissues by IHC. SIRT1 protein was expressed in both endometrial glandular and stromal cells. In the epithelial cells, SIRT1 level was increased in both the eutopic and ectopic endometrium of endometriotic patients, compared with normal endometrium (p < 0.0001, for both), but its levels in the eutopic and ectopic endometrium of endometriotic patients did not differ (p = 0.67) (Fig. 5C,D). Accordingly, the PCR and western blot results showed that SIRT1 expression was upregulated in ectopic endometrial tissues compared with normal control endometrial tissues (Fig. 5E,F).
Overexpression of SIRT1 allows endometrial epithelial cells (EECs) to evade senescence
To confirm the activity of SIRT1 in endometrial epithelial cells, resveratrol (RSV) as a SIRT1 activator and siRNA targeting SIRT1 were used in the Ishikawa cell line. qRT–PCR analysis showed that SIRT1 mRNA content was reduced by 49% and that RSV reversed the downregulation of SIRT1 in response to siRNA (Fig. 6A). RSV restored whereas knockdown of SIRT1 promoted cellular senescence in Ishikawa cells as ascertained by SAβG (Fig. 6B). As expected, SIRT1 knockdown was associated with an increase in the expression of p53 and p38MAPK, and RSV abolished the upregulation of senescence-associated genes in Ishikawa cells (Fig. 6C,D). Furthermore, immunofluorescence staining showed that SIRT1 knockdown induced p53 accumulation and downregulation of p53 in response to RSV (Fig. 6E). The data indicate that the upregulation of SIRT1 induced by RSV is SIRT1- dependent and allows cells to escape senescence.

Effect of RSV on the senescence of Ishikawa cells. qPCR analysis of SIRT1 mRNA levels in Ishikawa cells first transfected with siNC or siSIRT1 and then treated with or without RSV for 24 h (A). Representative SaβG staining in parallel Ishikawa cell cultures (B). qPCR analysis of p53 and p38 mRNA levels in Ishikawa cells first transfected with siNC or siSIRT1 and then treated with or without RSV for 24 h (C). Representative western blot and quantification of senescence-related genes from parallel cell cultures (D). p53 and SIRT1 protein levels were examined by immunofluorescence staining (E). RSV resveratrol. Scale bars = 50 µm in (E). *p < 0.05, **p < 0.01, ***p < 0.001.
Overexpression of SIRT1 promotes epithelial cells (EECs) EMT
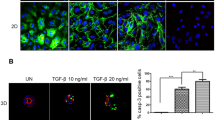
Since SIRT1 has been shown to promote EMT and favour the metastatic process in endometrial cancer24, we next examined the features of EMT in the activation or knockdown of SIRT1 in Ishikawa cells. Consistent with SIRT1 knockdown-mediated cellular senescence, a significant increase in the E-cadherin epithelial marker and a decrease in the vimentin mesenchymal marker were observed in Ishikawa cells. In contrast, RSV triggered a partial loss of E-cadherin, an increase in vimentin expression (Fig. 7A–C) and the acquisition of migration and invasive properties (Fig. 7D and Supplementary Fig. S7). The regulation of SIRT1 signalling through the interplay between senescence and EMT in endometrial epithelial cells is mapped in Fig. 7E. Together, these data suggest that overexpression of SIRT1 allows Ishikawa cells to override cancer-associated senescence and promote EMT, thereby facilitating endometriosis migration and invasion.

Effect of RSV on the EMT of Ishikawa cells. Ishikawa cells were treated with siNC or siSIRT1 with or without RSV, and then E-cad protein levels were examined by immunofluorescence staining (A). E-cad and vimentin mRNA or protein levels were examined by qPCR (B) and western blotting (C). Matrigel invasion assays to determine the invasive properties of the cells from the parallel cell cultures (D). The bioinformatics analysis depicting the correlation between SIRT1, ECM and senescence was mapped with the STRING database to construct a PPI network (E). Scale bars = 50 µm in (A). RSV resveratrol, E-cad E-cadherin. *p < 0.05, **p < 0.01, ***p < 0.001.
Discussion
In the current study we demonstrated that two important processes, senescence and EMT are involved in endometriosis. In endometrial epithelial cells, SIRT1 overrides the senescence program and contributes to the induction of EMT and the acquisition of metastatic capacity.
EMT represents a biological program during which epithelial cells lose their cellular identity and acquire a mesenchymal phenotype. This process is associated with the acquisition of tissue invasiveness. Epithelial-mesenchymal plasticity promotes the spread of ectopic lesions and can be activated by a number of signaling pathway in endometriosis33,43. Multiple cancer-related genes are believed to be EMT drivers. However, while it has been postulated that the endometrial epithelial cells carried distinct cancer-associated alterations that may be more susceptible to endometriosis44,45, it is still an open question of how EMT is triggered during endometriosis. Furthermore, the intimate crosstalk between EMT and the failsafe program has been confirmed16, but it is not clear whether SIRT1 regulates EMT, senescence and the capacity of metastasis in endometrial epithelial cells. Accordingly, its upregulated expression is associated with the pathogenesis of endometriosis.
Recent studies have shown that senescent cells are present in different types of tissues, including the human endometrium22. Consistent with our current study, it could be suggested that the senescent endometrial epithelial cells share features of damage-induced senescence, including the increased SAβG activity and the expression of senescence-related markers (p53, p16 and p38MAPK)22. In addition, telomere shortening has been established as one of the major mechanisms of aging46. Ectopic endometriotic lesions have aberrant telomerase and longer telomeres in the epithelial cells47, suggesting that epithelial cells may escape senescence in endometriosis. Indeed, several signalling pathways have been found to be altered during trancriptome analysis of endometria from endometriosis. Among these are the p53, RAS (rat sarcoma virus) and MAPK-dependent pathways4. The role of SIRT1 in endometriosis was previously demonstrated in mice and humans, which suggests that an increase in SIRT1 expression is associated with oncogene KRAS activation and contributes to progesterone resistance in endometriosis27,48. Importantly, SIRT1 is an essential factor that delays cellular senescence by regulating diverse biological processes by deacetylating transcription factors, including p53, forkhead transcription factors (FOXOs), p38MAPK and SMADs49. Therefore, endometrial epithelial cells retain the potential to escape senescence by mechanisms that involve ectopic SIRT1 overexpression in endometriosis. Our results showed that SIRT1 was overexpressed in endometriosis and that its induction is sufficient to override damage induced senescence in epithelial cells by inhibiting both the p53 and p38MAPK pathways. It has been KRAS can activate senescence-like growth arrest, whereas SIRT1 overexpression inhibits KRAS-induced senescence and SIRT1 knowdown reduces ability of SIRT1 to prevent KRAS-induced senescence50, which was consistent with our results. Although it has been suggested that pharmacological inhibition of SIRT1 induces senescence or apoptosis, it is difficult to rule out off-target effects due to the limited specificity of these compounds51. Therefore, our hypothesis was further verified by the overexpression and knockdown of SIRT1. We demonstrated that, the inverse correlation between p53, p38 MAPK and SIRT1 protein in the endometrium also supports the role of SIRT1 protein as a negative modulator of p53 and p38 MAPK, as a crucial inhibitor of damage-induced senescence in human endometrial epithelial cells. One of the limitations is that we employed Ishikawa human endometrial adenocarcinoma cell line as a model of endometrial epithelial cells. Using immortalized endometrotic epithelial cell lines may provide a more convincing model for studying endometriosis in the future. SIRT1 is implicated in EMT activation, and its function in DNA repair and cell cycle control was found to be similar to that of a tumor promoter gene50,51. The best characterized event occurring in EMT is loss of E-cadherin. SIRT1 can deacetylate histone H3 and cause repression of E-cadherin50. Furthermore, a high frequency of SIRT1 overexpression was reported during tumor progression in endometrial carcinoma24,52. Noticeably, endometriosis shares many features with malignancy, such as EMT activation33. Indeed, activation of EMT is linked to the suppression of cellular senescence and escape from senescence is a general prerequisite for malignant conversion46. Further characterization at the molecular level showed that alterations in the p53- and p38MAPK-dependent pathway promote EMT53. Alternatively, the EMT in endometriotic epithelial cells might be driven by ZEB (zinc finger E-box binding)40,54. ZEB1 is required for SIRT1 recruitment to repress E-cadherin and EMT51. Downregulation of ZEB1 or ZEB2 triggers senescence both in vivo and in vitro via p53 or p21 activation19,55,56. Moreover, ZEB1 and ZEB2 expression are associated with SIRT1 overexpression and have been implicated in the suppression of p53 and p38 MAP49,57. Thus, EMT-competent cells may be capable to negating senescence checkpoint functions through ZEB1 and ZEB2, whereas cellular senescence may prevent SIRT1 from inducing ZEB to activate the EMT program.
In conclusion, we used integrated systemic biology analysis and found that enrichment of stromal fibroblasts and the potential activation of EMT are features of endometriosis tissues. Furthermore, the endometriotic lesions are characterized by significant overexpression of SIRT1 in endometrial epithelial cells, which is involved in triggering the EMT switch by escaping damage or senescence during progression of the pathology. One the limitations in endometrosis is This observation supports that genetic and epigenetic incident favors endometrial epithelia cells escape from senescence and fuel EMT process in endometriosis, what could be overcome by downregulation of SIRT1. Although further work is warranted in clarifying the off-target effects51, the development of SIRT1 modulators with enhanced potency and selectivity will be useful for treating endometriosis patients.
Methods
Public datasets
We used “endometrosis” as a keyword in the Gene Expression Omnibus (GEO) database, and four mRNA transcription datasets were collected, including GSE11691 (n = 9), GSE7305 (n = 10), GSE5108, (n = 11), and E-MEXP-1251(n = 12). The raw data were downloaded from the corresponding databases (https://www.ncbi.nlm.nih.gov/geo, and https://www.ebi.ac.uk/arrayexpress/) and analysed using the limma package (version 3.12) in R software (version 3.61), and genes with p value < 0.05 and Log2 (fold change) > 1 were considered DEGs.
Data analysis
Gene Ontology (GO) and KEGG pathway enrichment analysis for DEGs were performed using the “clusterProfiler” package (version: 3.13) of R (http://bioconductor.org/packages/release/bioc/html/clusterProfiler.html). KEGG and HALLMARK gene sets were used from the Molecular Signatures Database (MSigDB). Gene set enrichment analysis (GSEA) was implemented by version 4.1.0, and the enrichment results were interpreted by normalized enrichment score9 and a false discovery rate (FDR) value < 0.05.
The protein–protein interaction network of the chosen genes was constructed by STRING (https://string-db.org, version 11.5), with a score > 0.400 corresponding to a median confidence interaction as significant.
To dissect the abundance of cell types in endometrial tissues, the xCell package (version: 1.36.0) (https://github.com/dviraran/xCell) was used to explore the tissue cellular heterogeneity from the gene expression data.
Clinical sample collection
This study was performed in accordance with the ethical principles outlined by the Helsinki Declaration and was approved by the Clinic Trial Ethics Committee of Longgang District People’s Hospital, Shenzhen, China (approval NO. 2022012). After informed consent, endometriosis subjects were recruited from women presenting with symptoms and subsequently confirmed by laparoscopic surgery and postoperative histological examination. Staging was performed according the revised American Fertility Society (r-AFS) classification into minimal-mild (stage I/II) and moderate-severe (III/IV) groups. Healthy fertile subjects were recruited from patients undergoing interval tubectomy and classified as controls. All subjects with age ≥ 40 years, ovarian tumors, polycystic ovarian syndrome, anatomical uterine abnormalities, pelvic inflammatory diseases and other pelvic pathological conditions were excluded. Additionally, all involved subjects had not received any hormonal treatment and did not use an intrauterine contraceptive device in the last 3 months prior to examination and sampling. Blood and tissue samples were collected during the proliferative phase of the menstrual cycle, which was determined by preoperative history and histological examination. The patient details are shown in Table 2.
Immunohistochemistry and immunofluorescence microscopy
Tissues were fixed, embedded in paraffin, sectioned (5 µm thick) and stained with haematoxylin and eosin (H&E) and Masson’s trichrome. The methods was performed as previously described57. Antibodies were used against the following targets: β-galactosidase (β-gal) (1:100 dilution, Santa Cruz, USA), p16 (1:100 dilution, CST, USA), p38 (1:50 dilution, Abcam, USA), E-cadherin (1:100 dilution, BD Biosciences, USA), vimentin (1:100 dilution, CST, USA) and SIRT1 (1:200 dilution, Abcam, USA). For the negative control, the primary antibody was replaced with an isotype control antibody. Immunohistochemistry and immunofluorescent staining and semiquantitative analysis by immunohistochemical reactive scores (IRSs) were conducted as previously described. Photographs were taken using an Olympus IX51epi-fluorescence microscope.
RT-qPCR and western blotting analysis
RT–qPCR and western blotting were performed as previously described57. The specific primers and antibodies are presented in Supplementary Table S2. The expression of genes was normalized to GAPDH. The results are representative of three independent experiments.
Cell culture and RNA interference
Ishikawa cells (BNCC, China) were cultured in DMEM/F12 (HyClone, USA) supplemented with 5% fetal bovine serum (FBS) (5% CO2, 20% O2, 37 °C). For the experiments, subconfluent Ishikawa cells were incubated in resveratrol (RSV) (Selleck, USA) for 24 h. The siRNAs against human SIRT1 (siSIRT1) and a negative control (siNC) were purchased from Guangzhou RiboBio Co., Ltd. (Guangzhou, China), and the sequences are presented in Supplementary Table S2. Cells were transfected with siRNA using Lipofectamine according to the manufacturer’s instructions (Invitrogen, USA).
SAβG staining
Senescent cells were detected using the Senescence β-Galactosidase Staining Kit (Beyotime, China) and observed under a phase-contrast microscope57. The quantification of positive cells was performed by counting the cells in 5 random fields per dish. At least 300 cells were randomly counted from each sample.
Transwell assay
A total of 5 × 104 cells in 100 µL serum-free DMEM/F-12 were seeded into the upper compartment of a 24-well chamber precoated with Matrigel (BD Biosciences, USA). The lower chamber was filled with DMEM/F 12 containing 10% FBS. After 24 h of incubation, cells on the upper surface of the filter were removed by wiping with a wet cotton swab and it was washed several times with PBS. The migrated cells on the lower surface of the upper chamber were fixed with 4% paraformaldehyde, stained with 0.1% crystal violet, and counted in five random fields under a light microscope.
Statistical analysis
GraphPad Prism software 8.0 was used to analyse the gene expression data generated from the IHC, qPCR and western blotting. Statistical significance was defined as p < 0.05. For single comparisons, unpaired t-tests were performed. One-way analysis of variance (ANOVA) was used for multiple comparisons. The results are presented as the mean ± SD for normally distributed data or as the median and interquartile range for nonnormally distributed data.
Data availability
The GSE11691, GSE7305 and GSE5108 datasets analyzed during the current study are available in the Gene Expression Omnibus data base, [https://www.ncbi.nlm.nih.gov/geo/]. The E-MEXP-1251 dataset analyzed during the current study are available in the Array Express, [https://www.ebi.ac.uk/arrayexpress/experiments/E-MEXP-1251/].
References
Gordts, S., Koninckx, P. & Brosens, I. Pathogenesis of deep endometriosis. Fertil. Steril. 108, 872–885. https://doi.org/10.1016/j.fertnstert.2017.08.036 (2017).
Guo, S. W. Cancer-associated mutations in endometriosis: Shedding light on the pathogenesis and pathophysiology. Hum. Reprod. Update. 26, 423–449. https://doi.org/10.1093/humupd/dmz047 (2020).
Guo, S. W. Fibrogenesis resulting from cyclic bleeding: The Holy Grail of the natural history of ectopic endometrium. Hum. Reprod. 33, 353–356. https://doi.org/10.1093/humrep/dey015 (2018).
Chen, M. et al. Bioinformatic analysis reveals the importance of epithelial-mesenchymal transition in the development of endometriosis. Sci. Rep. 10, 8442. https://doi.org/10.1038/s41598-020-65606-9 (2020).
Poli-Neto, O. B., Meola, J., Rosa-E-Silva, J. C. & Tiezzi, D. Transcriptome meta-analysis reveals differences of immune profile between eutopic endometrium from stage I-II and III-IV endometriosis independently of hormonal milieu. Sci. Rep. 10, 313. https://doi.org/10.1038/s41598-019-57207-y (2020).
Ma, L. et al. Epithelial-to-mesenchymal transition contributes to the downregulation of progesterone receptor expression in endometriosis lesions. J. Steroid Biochem. Mol. Biol. 212, 105943. https://doi.org/10.1016/j.jsbmb.2021.105943 (2021).
Proestling, K. et al. Enhanced epithelial to mesenchymal transition (EMT) and upregulated MYC in ectopic lesions contribute independently to endometriosis. Reprod. Biol. Endocrinol. 13, 75. https://doi.org/10.1186/s12958-015-0063-7 (2015).
Zeitvogel, A., Baumann, R. & Starzinski-Powitz, A. Identification of an invasive, N-cadherin-expressing epithelial cell type in endometriosis using a new cell culture model. Am. J. Pathol. 159, 1839–1852. https://doi.org/10.1016/S0002-9440(10)63030-1 (2001).
Anglesio, M. S. et al. Cancer-associated mutations in endometriosis without cancer. N. Engl. J. Med. 376, 1835–1848. https://doi.org/10.1056/NEJMoa1614814 (2017).
Yu, C. X. et al. Correlation between replicative senescence of endometrial gland epithelial cells in shedding and non-shedding endometria and endometriosis cyst during menstruation. Gynecol. Endocrinol. 34, 981–986. https://doi.org/10.1080/09513590.2018.1480709 (2018).
Risques, R. A. & Kennedy, S. R. Aging and the rise of somatic cancer-associated mutations in normal tissues. PLoS Genet. 14, e1007108. https://doi.org/10.1371/journal.pgen.1007108 (2018).
Alnafakh, R. et al. Endometriosis is associated with a significant increase in hTERC and altered telomere/telomerase associated genes in the eutopic endometrium, an ex-vivo and in silico study. Biomedicines. 8, 588. https://doi.org/10.3390/biomedicines8120588 (2020).
Parvanov, D., Ganeva, R., Vidolova, N. & Stamenov, G. Decreased number of p16-positive senescent cells in human endometrium as a marker of miscarriage. J. Assist. Reprod. Genet. 38, 2087–2095. https://doi.org/10.1007/s10815-021-02182-5 (2021).
Luo, M., Cai, X., Yan, D., Liu, X. & Guo, S. W. Sodium tanshinone IIA sulfonate restrains fibrogenesis through induction of senescence in mice with induced deep endometriosis. Reprod. Biomed. Online. 41, 373–384. https://doi.org/10.1016/j.rbmo.2020.04.006 (2020).
Ansieau, S. et al. Induction of EMT by twist proteins as a collateral effect of tumor-promoting inactivation of premature senescence. Cancer Cell 14, 79–89. https://doi.org/10.1016/j.ccr.2008.06.005 (2008).
Wen, F. C., Chang, T. W., Tseng, Y. L., Lee, J. C. & Chang, M. C. hRAD9 functions as a tumor suppressor by inducing p21-dependent senescence and suppressing epithelial-mesenchymal transition through inhibition of Slug transcription. Carcinogenesis 35, 1481–1490. https://doi.org/10.1093/carcin/bgu009 (2014).
Liu, W. & Sharpless, N. E. Senescence-escape in melanoma. Pigment Cell Melanoma Res. 25, 408–409. https://doi.org/10.1111/j.1755-148x.2012.01021.x (2012).
Kratz, E. M., Kokot, I., Dymicka-Piekarska, V. & Piwowar, A. Sirtuins-the new important players in women’s gynecological health. Antioxidants 10, 84. https://doi.org/10.3390/antiox10010084 (2021).
Liu, Y., El-Naggar, S., Darling, D. S., Higashi, Y. & Dean, D. C. Zeb1 links epithelial-mesenchymal transition and cellular senescence. Development 135, 579–588. https://doi.org/10.1242/dev.007047 (2008).
Smit, M. A. & Peeper, D. S. Deregulating EMT and senescence: Double impact by a single twist. Cancer Cell 14, 5–7. https://doi.org/10.1016/j.ccr.2008.06.012 (2008).
Velarde, M. C. & Menon, R. Positive and negative effects of cellular senescence during female reproductive aging and pregnancy. J. Endocrinol. 230, R59–R76. https://doi.org/10.1530/JOE-16-0018 (2016).
Brighton, P. J. et al. Clearance of senescent decidual cells by uterine natural killer cells in cycling human endometrium. Elife 6, e31274. https://doi.org/10.7554/eLife.31274 (2017).
Hashimoto, M. et al. Analysis of telomeric single-strand overhang length in human endometrial cancers. FEBS Lett. 579, 2959–2964. https://doi.org/10.1016/j.febslet.2005.04.021 (2005).
Asaka, R. et al. Sirtuin 1 promotes the growth and cisplatin resistance of endometrial carcinoma cells: A novel therapeutic target. Lab. Invest. 95, 1363–1373. https://doi.org/10.1038/labinvest.2015.119 (2015).
Kim, T. H. et al. Role of SIRT1 and progesterone resistance in normal and abnormal endometrium. J. Clin. Endocrinol. Metab. https://doi.org/10.1210/clinem/dgab753 (2021).
Sansone, A. M. et al. Evaluation of BCL6 and SIRT1 as non-invasive diagnostic markers of endometriosis. Curr. Issues Mol. Biol. 43, 1350–1360. https://doi.org/10.3390/cimb43030096 (2021).
Yoo, J. Y. et al. KRAS activation and over-expression of SIRT1/BCL6 contributes to the pathogenesis of endometriosis and progesterone resistance. Sci. Rep. 7, 6765. https://doi.org/10.1038/s41598-017-04577-w (2017).
Teasley, H. E. et al. Differential expression of KRAS and SIRT1 in ovarian cancers with and without endometriosis. Reprod. Sci. 27, 145–151. https://doi.org/10.1007/s43032-019-00017-4 (2020).
Zheng, J., Shao, S., Dai, C., Guan, S. & Chen, H. miR-9-5p promotes the invasion and migration of endometrial stromal cells in endometriosis patients through the SIRT1/NF-kappaB pathway. Int. J. Clin. Exp. Pathol. 13, 1859–1866 (2020).
Rezk, N. A., Lashin, M. B. & Sabbah, N. A. MiRNA 34-a regulate SIRT-1 and Foxo-1 expression in endometriosis. Non-Coding RNA Res. 6, 35–41. https://doi.org/10.1016/j.ncrna.2021.02.002 (2021).
Taguchi, A. et al. Resveratrol suppresses inflammatory responses in endometrial stromal cells derived from endometriosis: a possible role of the sirtuin 1 pathway. J. Obstet. Gynaecol. Res. 40, 770–778. https://doi.org/10.1111/jog.12252 (2014).
Zhang, L., Li, H. H., Yuan, M., Li, D. & Wang, G. Y. Exosomal miR-22–3p derived from peritoneal macrophages enhances proliferation, migration, and invasion of ectopic endometrial stromal cells through regulation of the SIRT1/NF-kappaB signaling pathway. Eur. Rev. Med. Pharmacol. Sci. 24, 571–580. https://doi.org/10.26355/eurrev_202001_20033 (2020).
Kong, X. et al. MTA1, a target of resveratrol, promotes epithelial-mesenchymal transition of endometriosis via ZEB2. Mol. Ther. Methods Clin. Dev. 19, 295–306. https://doi.org/10.1016/j.omtm.2020.09.013 (2020).
Khodarahmian, M. et al. A randomized exploratory trial to assess the effects of resveratrol on VEGF and TNF-alpha 2 expression in endometriosis women. J. Reprod. Immunol. 143, 103248. https://doi.org/10.1016/j.jri.2020.103248 (2021).
Chen, Z. et al. Lipidomic alterations and PPARalpha activation induced by resveratrol lead to reduction in lesion size in endometriosis models. Oxid. Med. Cell Longev. 2021, 9979953. https://doi.org/10.1155/2021/9979953 (2021).
Wang, C. et al. Transcriptome-based analysis reveals therapeutic effects of resveratrol on endometriosis in arat model. Drug Des. Devel. Ther. 15, 4141–4155. https://doi.org/10.2147/DDDT.S323790 (2021).
Wei, Z. et al. Knockdown hsa_circ_0063526 inhibits endometriosis progression via regulating the miR-141–5p/EMT axis and downregulating estrogen receptors. Aging 13, 26095–26117. https://doi.org/10.18632/aging.203799 (2021).
Xiong, W. et al. E2 -mediated EMT by activation of beta-catenin/Snail signalling during the development of ovarian endometriosis. J. Cell Mol. Med. 23, 8035–8045. https://doi.org/10.1111/jcmm.14668 (2019).
Cela, V. et al. Exploring epithelial-mesenchymal transition signals in endometriosis diagnosis and in vitro fertilization outcomes. Biomedicines. 9, 1681. https://doi.org/10.3390/biomedicines9111681 (2021).
Zhang, Q., Dong, P., Liu, X., Sakuragi, N. & Guo, S. W. Enhancer of Zeste homolog 2 (EZH2) induces epithelial-mesenchymal transition in endometriosis. Sci. Rep. 7, 6804. https://doi.org/10.1038/s41598-017-06920-7 (2017).
Chatterjee, K., Jana, S., DasMahapatra, P. & Swarnakar, S. EGFR-mediated matrix metalloproteinase-7 up-regulation promotes epithelial-mesenchymal transition via ERK1-AP1 axis during ovarian endometriosis progression. FASEB J. 32, 4560–4572. https://doi.org/10.1096/fj.201701382RR (2018).
Chang, L. C. et al. The potential effect of fucoidan on inhibiting epithelial-to-mesenchymal transition, proliferation, and increase in apoptosis for endometriosis treatment: in vivo and in vitro study. Biomedicines. 8, 528. https://doi.org/10.3390/biomedicines8110528 (2020).
Yang, Y. M. & Yang, W. X. Epithelial-to-mesenchymal transition in the development of endometriosis. Oncotarget 8, 41679–41689. https://doi.org/10.18632/oncotarget.16472 (2017).
Suda, K. et al. Clonal expansion and diversification of cancer-associated mutations in endometriosis and normal endometrium. Cell Rep. 24, 1777–1789. https://doi.org/10.1016/j.celrep.2018.07.037 (2018).
Moore, L. et al. The mutational landscape of normal human endometrial epithelium. Nature 580, 640–646. https://doi.org/10.1038/s41586-020-2214-z (2020).
Patel, P. L., Suram, A., Mirani, N., Bischof, O. & Herbig, U. Derepression of hTERT gene expression promotes escape from oncogene-induced cellular senescence. Proc. Natl. Acad. Sci. U. S. A. 113, E5024–E5033. https://doi.org/10.1073/pnas.1602379113 (2016).
Valentijn, A. J., Saretzki, G., Tempest, N., Critchley, H. O. & Hapangama, D. K. Human endometrial epithelial telomerase is important for epithelial proliferation and glandular formation with potential implications in endometriosis. Hum. Reprod. 30, 2816–2828. https://doi.org/10.1093/humrep/dev267 (2015).
Evans-Hoeker, E. et al. Endometrial BCL6 overexpression in eutopic endometrium of women with endometriosis. Reprod. Sci. 23, 1234–1241. https://doi.org/10.1177/1933719116649711 (2016).
Hishida, T. et al. Sirt1, p53, and p38(MAPK) are crucial regulators of detrimental phenotypes of embryonic stem cells with Max expression ablation. Stem Cells. 30, 1634–1644. https://doi.org/10.1002/stem.1147 (2012).
Zhao, G. et al. SIRT1 RNAi knockdown induces apoptosis and senescence, inhibits invasion and enhances chemosensitivity in pancreatic cancer cells. Gene Ther. 18(9), 920–928. https://doi.org/10.1038/gt.2011.81 (2011).
Hu, J., Jing, H. & Lin, H. Sirtuin inhibitors as anticancer agents. Future Med. Chem. 6(8), 945–966. https://doi.org/10.4155/fmc.14.44 (2014).
Sun, T., Jiao, L., Wang, Y., Yu, Y. & Ming, L. SIRT1 induces epithelial-mesenchymal transition by promoting autophagic degradation of E-cadherin in melanoma cells. Cell Death. Dis. 9, 136. https://doi.org/10.1038/s41419-017-0167-4 (2018).
Byles, V. et al. SIRT1 induces EMT by cooperating with EMT transcription factors and enhances prostate cancer cell migration and metastasis. Oncogene 31, 4619–4629. https://doi.org/10.1038/onc.2011.612 (2012).
de Barrios, O. et al. ZEB1-induced tumourigenesis requires senescence inhibition via activation of DKK1/mutant p53/Mdm2/CtBP and repression of macroH2A1. Gut 66, 666–682. https://doi.org/10.1136/gutjnl-2015-310838 (2017).
Ohashi, S. et al. Epidermal growth factor receptor and mutant p53 expand an esophageal cellular subpopulation capable of epithelial-to-mesenchymal transition through ZEB transcription factors. Cancer Res. 70, 4174–4184. https://doi.org/10.1158/0008-5472.CAN-09-4614 (2010).
Xiao, Y. et al. p38/p53/miR-200a-3p feedback loop promotes oxidative stress-mediated liver cell death. Cell Cycle 14, 1548–1558. https://doi.org/10.1080/15384101.2015.1026491 (2015).
Wang, Y. et al. SIRT1 regulates trophoblast senescence in premature placental aging in preeclampsia. Placenta 122, 56–65. https://doi.org/10.1016/j.placenta.2022.04.001 (2022).
Acknowledgements
This work was supported by a National Natural Science Foundation of China (NSFC 81971395).
Author information
Authors and Affiliations
Contributions
M.W. recruited the patients and obtained specimens, performed the experiments together with J.L., Y.W. and J.H. Y.H. analysed and interpreted the data together with G.Q.; Y.W. and Y.C. prepared the figures; P.L. conceived and wrote the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information







Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, M., Wu, Y., He, Y. et al. SIRT1 upregulation promotes epithelial-mesenchymal transition by inducing senescence escape in endometriosis. Sci Rep 12, 12302 (2022). https://doi.org/10.1038/s41598-022-16629-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-16629-x
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}