
Abstract
Plants produce toxic secondary metabolites as defense mechanisms against phytopathogenic microorganisms and predators. L-azetidine-2-carboxylic acid (AZC), a toxic proline analogue produced by members of the Liliaceae and Agavaciae families, is part of such a mechanism. AZC causes a broad range of toxic, inflammatory and degenerative abnormalities in human and animal cells, while it is known that some microorganisms have evolved specialized strategies for AZC resistance. However, the mechanisms underlying these processes are poorly understood. Here, we identify a widespread mechanism for AZC resistance in fungi. We show that the filamentous ascomycete Aspergillus nidulans is able to not only resist AZC toxicity but also utilize it as a nitrogen source via GABA catabolism and the action of the AzhA hydrolase, a member of a large superfamily of detoxifying enzymes, the haloacid dehalogenase-like hydrolase (HAD) superfamily. This detoxification process is further assisted by the NgnA acetyltransferase, orthologue of Mpr1 of Saccharomyces cerevisiae. We additionally show that heterologous expression of AzhA protein can complement the AZC sensitivity of S. cerevisiae. Furthermore, a detailed phylogenetic analysis of AzhA homologues in Fungi, Archaea and Bacteria is provided. Overall, our results unravel a widespread mechanism for AZC resistance among microorganisms, including important human and plant pathogens.
Similar content being viewed by others

Introduction
L-Azetidine-2-carboxylic acid (AZC) is a toxic analogue of L-proline (L-Pro) produced by members of the Liliaceae, Agavaceae, Asparagaceae, Fabaceae families and by several Beta vulgaris cultivars, such as beetroot or sugar beet1,2,3. Although it is not fully understood why some plants produce toxic compounds like AZC, it has been suggested that such compounds can serve as a defense mechanism, by poisoning predators, pathogens or competitors, thus protecting against infections and consumption, or impeding the growth of neighboring plants4,5,6. AZC defense mechanism relies on the stereochemical similarity of the non-proteinogenic imino acid AZC with the proteinogenic L-Pro, which differ by only one C atom7. AZC is incorporated into proteins during protein synthesis, as it can be loaded on L-proline-tRNA instead of L-Pro, a phenomenon that has been characterized as protein misincorporation or amino acid mimicry8,9,10. With one C atom less, AZC is less flexible than L-Pro, resulting in polypeptides with a capability of rotation reduced by 15°8,11. These changes eventually affect the folding and the tertiary structure of the proteins in which AZC is incorporated, resulting in non-functional, misfolded proteins. The accumulation of these protein products becomes deleterious and, finally, obstructs cell proliferation and growth12,13,14.
Mechanisms of resistance to AZC have been described in plants, fungi and bacteria15. It has been suggested that AZC-producing plants protect themselves from AZC auto-toxicity, by avoiding the loading of AZC on L-Pro-tRNA via at least three mechanisms: (a) their L-Pro-tRNA activating enzymes are highly specific and discriminate between L-pro and AZC (b) AZC is subcellularly excluded from protein-synthesis sites16 and (c) some plants possess an efficient tRNA synthetase editing system, able to remove AZC loaded on L-Pro-tRNA9.
In fungi, it has long been shown that some strains of the budding yeast Saccharomyces cerevisiae with increased intracellular levels of L-Pro are resistant to AZC17. Moreover, a specific strain, Σ1278b, was shown to detoxify AZC through the function of Mpr1/2 acetyltransferases18. Mpr1/2, members of the N-acetyl-transferase superfamily, were shown to catalyze the acetylation of AZC to produce N-acetyl-AZC, a compound that is no longer recognized by tRNA synthetases and is therefore non-toxic to the cell19. However, AZC detoxifying activity of Mpr1/2 has been so far identified only in the Σ1278b strain of S. cerevisiae and in very few other yeast species20,21.
In bacteria, an additional mechanism of AZC detoxification has been described in a Pseudomonas strain, which was isolated from soil surrounding the roots of the AZC-producing plant Convallaria majalis, a member of the Liliaceae family. This bacterium was found to possess a hydrolase (AC-hydrolase) able to detoxify AZC22. The underlying molecular mechanism leads to the periplasmic hydrolysis of AZC to 2-hydroxy-4-aminobutyrate, a substance that could then be transported into the cytoplasm by the action of a GABA transporter homologue, where it subsequently undergoes transamination22.
In order for AZC to be toxic, its intracellular accumulation is essential. In yeast, this is known to occur via active transport by three broad specificity amino acid transporters (Agp1, Gnp1 and the general amino acid permease, Gap1) as well as the specialized L-Pro transporter, Put423. In a previous report, we described the specificity of PrnB, the Put4 orthologue in A. nidulans, and we demonstrated that, contrary to Put4, PrnB is highly specific to L-Pro and does not recognize AZC. Remarkably, during this work we noticed that A. nidulans shows resistance to AZC, for which its Mpr1 orthologue NgnA (formerly recorded as Ngn224) is not necessary25.
In the present study, we characterize the mechanism underlying resistance of potentially several fungi to AZC, using A. nidulans as a model organism. We report that A. nidulans possesses the ability not only to detoxify AZC, but also to utilize it as a poor nitrogen source, by its catabolism via the GABA pathway25,26,27. We show that AzhA, an AC-hydrolase orthologue, is necessary and sufficient for AZC detoxification and assimilation. An in silico analysis revealed that AzhA is a member of the haloacid dehalogenase-like hydrolase (HAD) super-family, a large group of enzymes with diverse substrate specificity that has been associated with the detoxification of toxic compounds and xenobiotics28. Furthermore, we report that NgnA acetyltransferase is involved in AZC detoxification and we provide a detailed phylogenetic analysis of AzhA amino acid sequence across the three domains of life.
Results
AZC detoxification and assimilation in A. nidulans
Consistently with our previous results25, we confirmed that AZC is not toxic to A. nidulans. In this study, we further demonstrated that AZC can be utilized as a sole nitrogen source (Fig. 1). Therefore, given that growth with AZC as sole nitrogen source is NgnA-independent (Fig. 1), we investigated whether an AZC-hydrolase, similar to the AZC-assimilating hydrolase of Pseudomonas A2C strain (AC-hydrolase), is present in A. nidulans. The Basic Local Alignment Search Tool for proteins (BLASTp), and the amino acid sequence of Pseudomonas AC-hydrolase as query (Fig. S1 in supplementary material), were used to retrieve sequences with strong similarity in A. nidulans. The results obtained, indicated the product of the AN12472 gene (designated also as AN9518) as a top-scoring match, with 44.54% percentage identity and 97% query coverage. AN12472 is an uncharacterized gene, with an intronless Open Reading Frame (ORF) of 726 bp, encoding a 241 amino acid polypeptide with a predicted hydrolase activity, acting on acid halide bonds. This amino acid sequence was designated AzhA (Αzetidine hydrolase). Deletion of the azhA gene resulted in the inability of the corresponding strain to utilize AZC, as evidenced by a noticeable decrease in the growth of the azhAΔ deletion strain in the presence of AZC as sole nitrogen source (Fig. 1A). In support of this, AZC is slightly toxic to azhAΔ in the presence of urea (Fig. 1A,B). As previously reported, deletion of the ngnA gene (encoding an MPR1/2 orthologue), does not confer AZC sensitivity to A. nidulans25. Moreover, ngnAΔ deletion strain macroscopically grows similarly to a wild-type (WT) strain in the presence of AZC as sole nitrogen source. Hence, we examined the growth of the azhAΔ ngnAΔ double deletion strain, in comparison to the WT and the corresponding single deletion strains, azhAΔ and ngnAΔ. As documented in Fig. 1A, growth of the azhAΔ ngnAΔ strain is severely impaired by the presence of AZC, when urea is used as a nitrogen source, supporting the involvement of both enzymes in AZC detoxification.

AzhA is required for growth of A. nidulans in the presence of AZC. (A) Growth of WT, azhAΔ, ngnAΔ, azhAΔ ngnAΔ, gatA-, amdR-, gabAΔ, prnBΔ, gabAΔ prnBΔ and prnA- A. nidulans strains on MM with Urea (U); AZC; GABA and L-Pro, as nitrogen sources at a final concentration of 5 mM, or on MM lacking any nitrogen source (–N). Growth of ngnAΔ single deletion strain in the presence of AZC either as sole nitrogen source (AZC) or with urea (AZC + U) is similar to that of the WT strain. On the other hand, growth of azhAΔ single deletion strain in the presence of AZC as sole nitrogen is severely impaired, while in AZC + U is similar to that of the WT, indicating that AzhA is involved in AZC assimilation. Accordingly, growth of the ngnAΔ azhAΔ double deletion strain is severely impaired in both AZC or in AZC + U, indicating the involvement of the corresponding enzymatic activities in AZC detoxification and assimilation. Growth of gatA- and amdR- strains on AZC as sole nitrogen source is highly reduced compared to the WT strain, supporting that AZC is assimilated through the GABA metabolic pathway. On the contrary, growth of prnBΔ and prnA- strains on AZC as sole nitrogen source is similar to that of the WT strain, supporting that AZC is not assimilated through the proline catabolic pathway. Finally, gabAΔ, prnBΔ, gabAΔ prnBΔ mutant strains show no growth defect in the presence of AZC as sole nitrogen source, suggesting that AZC is not transported through the major proline transporter PrnB or the GABA transporter GabA. (B) Growth rate of A. nidulans WT, azhAΔ, ngnAΔ and azhAΔ ngnAΔ germlings in submerged liquid cultures, using (U), (AZC + U) or AZC as nitrogen sources. Liquid media were inoculated with conidiospores and incubated for 18 h at 25 °C. Hyphal growth rate was observed at frames taken every 15 min for a total of 45 min. Values are plotted in box-and-whiskers plots. Asterisks indicate a significant difference of the mean (P ≤ 0.05) between two conditions. Statistical significance was analyzed via the Kruskal–Wallis Test and is depicted with asterisks (*). Single (*), double (**) or triple (***) asterisks, indicate 0.01 ≤ p < 0.05, 0.001 < P ≤ 0.01, or p < 0.001, respectively.
As a complementary to the above approach, we measured the hyphal growth rate of several strains in the presence or absence of AZC, in submerged liquid cultures, using confocal microscopy. The results (Fig. 1B, see also Video S1 in supplementary material) show that WT hyphae from germlings that have grown for 18 h with urea as sole nitrogen source, have an apical growth rate of 25.89 ± 10.38 μm/h. This rate is nearly identical (26.12 ± 14.96 μm/h) to that of germlings incubated in the simultaneous presence of urea and AZC (U + AZC), further confirming that AZC is not toxic to A. nidulans. Accordingly, azhAΔ and ngnAΔ single deletion strains have almost similar mean values of growth rate, when grown in urea. Interestingly, we noticed that the growth rate of the azhAΔ ngnAΔ double mutant germlings with urea as sole nitrogen source was statistically significantly lower compared to either the WT or the single deletion strains. This observation was also evident when different nitrogen sources were used, e.g. ammonium and glutamate (data not shown), suggesting that both enzymes might somehow be implicated in the hyphal polar growth or germination of conidiospores. As expected, the growth rate of the WT strain was reduced in the presence of AZC as the sole nitrogen source, with a mean growth velocity of 16.91 ± 13.97 μm/h, confirming that AZC is a poor nitrogen source for A. nidulans. On the contrary, the growth rate of the azhAΔ deletion strain significantly decreased when both AZC and urea were present in the growth medium. Growth of the azhAΔ ngnAΔ double mutant was, as expected, severely inhibited when AZC was supplemented as sole nitrogen source, in agreement with growth tests in solid media. When both urea and AZC were supplemented, only approximately 20% of cells accomplished emergence of the germ tube, indicating the severe toxicity of AZC in the double deletion strain (Fig. 1B).
In addition, it was observed that ngnAΔ single deletion strain in U + AZC had a growth rate 1.5 fold lower than in urea (U) alone. This phenotype of the ngnAΔ strain, which was not clearly observed with the conventional solid media growth tests, indicates an important contribution of the NgnA acetyltransferase in the detoxification of AZC in germlings. In total, the microscopic approach used for growth observations29,30, enabled us to measure more clearly the polar growth rate of our strains grown with different nitrogen sources and to further confirm that both AzhA and NgnA proteins contribute to AZC detoxification, while only AzhA is required for the utilization of AZC as a nitrogen source in A. nidulans.
Catabolism of AZC in A. nidulans is AmdR/IntA- and GABA-dependent and L-proline independent
In order to gain insights into the metabolic pathway of AZC catabolism in A. nidulans, and given that it is a proline analogue whose detoxified hydrolysis product in Pseudomonas is transported into the cytoplasm by the action of a GABA transporter homologue (see above), we examined AZC transport in strains defective for proline (prnBΔ) and γ-aminobutyric acid (GABA) (gabAΔ) uptake, as well as in strains defective in the GABA (amdR-, gatA-) or L-Pro (prnA-) metabolic pathways. The prnBΔ strain carries a deletion in the gene of the main proline transporter, PrnB31,32, and accordingly the gabAΔ carries a deletion in the gene coding for a GABA permease25,33. Strains designated as gatA- and amdR- carry loss-of-function mutations in the genes expressing the GABA transaminase (GatA), and its positively acting regulator AmdR/IntA, respectively26,27,34. Finally, the prnA- strain carries a loss-of-function mutation in the gene expressing the specific positively acting regulator of the five clustered structural genes of L-Pro uptake and catabolism35,36. Growth tests presented in Fig. 1A show that, in the presence of AZC as sole nitrogen source, both gatA- and amdR- loss-of-function strains have severely reduced growth compared to WT, similar to that of the azhAΔ strain. This reduction in growth is probably due to the inability of the gatA- and amdR- strains to sufficiently assimilate AZC via the GABA metabolic pathway, rather than AZC as a sole nitrogen source itself being toxic, since growth of both strains in AZC + U is similar to that of the WT strain. On the other hand, strains lacking either the main L-Pro (prnBΔ), or the main γ-aminobutyric (gabAΔ) transporters or both (prnBΔ gabAΔ), show no growth defect in the presence of AZC as sole nitrogen source (Fig. 1A), suggesting the existence of other transporter(s) responsible for AZC uptake. This transporter is not the product of AN5678, the strict homologue of the general amino acid permease Gap1 of S. cerevisiae37, since the triple deletion strain lacking all permeases is still able to utilize AZC (data not shown). Furthermore, the prnA- loss-of-function strain shows similar growth to the WT in the presence of AZC, indicating that AZC is not catabolized via the L-Pro catabolic pathway (Fig. 1A).
Expression of azhA and ngnA is induced by AZC
To our knowledge, there is little information on the regulation of expression of genes involved in resistance to AZC in S. cerevisiae, Pseudomonas or their homologues in plants15,38. Expression of A. nidulans genes encoding proteins involved in the uptake and catabolism of nitrogenous compounds is mainly regulated at the transcriptional level and is induced only when their substrates are available34,35,35. Therefore, we examined whether the expression of the azhA and ngnA genes is subject to transcriptional regulation by AZC, using RT-PCR. RNAs were extracted from a WT strain grown with urea (U) as a nitrogen source in the presence or absence of AZC. Our results show an approximately four-fold increase of azhA (Fig. 2A) and more than a 15-fold increase of ngnA transcript levels in the presence of AZC (Fig. 2A), indicating that AZC acts as a potent inducer of the genes involved in AZC detoxification and/or catabolism. Interestingly, we have observed that ngnA basal expression levels were quite low in non-induced conditions, and we could detect a slow migrating band corresponding to unspliced RNA (pre-mRNA), which we confirmed was not due to DNA contamination (see Materials and Methods). In the presence of AZC this band disappeared, suggesting an AZC-dependent induction in the splicing of the ngnA gene (investigation in progress).

Expression of azhA and ngnA genes in different strains and growth conditions. Total RNA was extracted from hyphae of WT, areA600, creAd and amdR-, strains on appropriately supplemented MM and incubated at 25 °C. (A) Representative images of RT-PCR-derived mRNA transcripts of azhA and ngnA genes in the presence of urea (-) or urea and AZC (AZC) as nitrogen sources and glucose as carbon source. (B and C) Effects of L-Proline (Pro), GABA or ammonium (NH+4) as nitrogen sources and glucose (Gl), fructose (Fr) or ethanol (EtOH) as carbon sources, on the expression of the azhA (B) and ngnA (C) genes, respectively in a WT strain. Levels of azhA and ngnA transcripts in different conditions were normalized by 18S rRNA levels. Statistical significance was analyzed via two-way ANOVA and is depicted with asterisks (*). Single (*), double (**) or triple (***) asterisks, indicate 0.01 ≤ p < 0.05, 0.001 < P ≤ 0.01, or p < 0.001, respectively.
Since AZC is an L-Pro analogue, we further investigated the involvement of L-Pro in the regulation of ngnA and azhA gene expression. It is well established that A. nidulans is able to utilize L-Pro as sole nitrogen and carbon source using enzymes mapped in five genes clustered in linkage group VII (prn cluster). The structural genes of the prn cluster are not transcribed in the absence of L-Pro, or in a genetic background lacking the cluster specific positive regulatory protein, PrnA41. Our results presented in Fig. 2B and C show that L-Pro does not induce the expression of either azhA or ngnA genes. Along with that, absence of PrnA does not affect either the utilization of AZC as poor nitrogen source or its toxicity, as evidenced by the similar growth of a prnA- loss-of-function and the WT strain in the presence of AZC (Fig. 1A).
In A. nidulans, the enzymes involved in the utilization of nitrogenous sources are subject to nitrogen metabolite repression (NMR), mediated by the general transcriptional activator AreA34,36. Thus, as AZC can be utilized as a poor nitrogen source (Fig. 1A), we have examined whether azhA and ngnA expression is regulated by AreA. Towards this, the expression of both genes was investigated in an areA null mutant strain (areA600) and compared to that of the WT strain, using RT-PCR. An areA600 strain favors ammonium as nitrogen source for its growth, while it shows a moderate growth with urea and a complete lack of growth with any other secondary nitrogen source, like L-Pro or nitrate42. Our results presented in Fig. 2A, show that in the presence of AZC, ngnA expression is reduced in areA600 compared to the WT strain, suggesting an AreA-dependent ngnA expression. However, AZC-induced ngnA expression is not affected by the presence of ammonium (Fig. 2C), a condition well-known to inactivate AreA36,43. Therefore, it appears that reduced induction of ngnA in areA600 is most probably due to an ammonium independent AreA regulation (see discussion). On the other hand, transcript levels of azhA remain approximately similar in WT and areA600 strains, suggesting an AreA-independent regulation of azhA expression (Fig. 2B). Interestingly, in an azhAΔ ngnAΔ double mutant strain ammonium partially protects from AZC toxicity (data not shown), suggesting an AreA-dependent expression of the AZC-transporter. This might in turn explain the small reduction observed in the AZC-induced levels of both the azhA and ngnA genes in the presence of ammonium.
In A. nidulans, the enzymes involved in the utilization of carbon sources, including L-Pro, are subject to carbon catabolite repression (CCR), mediated by the general transcriptional suppressor CreA. Therefore, as AZC is an L-Pro analogue, we have examined whether azhA and ngnA expression is regulated by CreA. Towards this, the expression of both genes was investigated in a constitutive carbon-catabolite-derepressed strain (creAd)44,45, by RT-PCR. As shown in Fig. 2A, azhA transcript levels are similar in creAd and WT strains, in both the presence and absence of AZC. On the contrary, ngnA transcript levels are slightly increased in creAd compared to the WT (Fig. 2A), in an AZC-independent manner. However, the basal levels of ngnA expression or the levels of its AZC-mediated induction are not increased when ethanol or fructose instead of glucose is utilized as a sole carbon source (Fig. 2B,C). As it is well established, CreA is inactive when a secondary carbon source like ethanol or fructose is utilized36.Thus, ngnA regulation by CreA is repressing carbon source independent.
Our data suggest that AZC is catabolised in A. nidulans through the GABA metabolic pathway (Fig. 1A). Hence, we investigated whether azhA and ngnA expression is regulated by GABA and/or AmdR/IntA, the positive acting specific transcription factor regulating the expression of five structural genes involved in acetamide (amdS), omega amino acid (gatA and gabA) and lactam (lamA and lamB) catabolism26,27,46. Firstly, transcript levels of both azhA and ngnΑ genes were examined, as above, in an amdR- null mutant strain. As shown in Fig. 2A, induction of azhA is similar in amdR- and WT strains. On the other hand, ngnA transcript levels were increased in the amdR- strain in the presence of AZC, suggesting that either AmdR/IntA acts as a transcriptional repressor of ngnA expression, or that a potential over-accumulation of AZC in the amdR- strain, due to lack of AZC assimilation, leads to overexpression of ngnΑ. The latter scenario would suggest that AZC itself, and not GABA is the inducer of azhA and ngnA expression. In order to investigate this possibility, we examined whether GABA acts as an inducer of azhA and ngnA expression. As documented in Fig. 2B,C, GABA does not induce either azhA or ngnA expression. Therefore, we conclude that AZC itself is the inducer of ngnA and azhA expression.
The above results are consistent with the possibility that AZC is converted to GABA in order to be assimilated. To examine this, we investigated whether AZC induces the expression of the gatA GABA transaminase gene, well-known to be induced by GABA and β-alanine27,33. As documented in Fig. 3A,B in the WT strain, gatA expression is induced in the presence of AZC in a way similar to the presence of β-alanine, its natural inducer40. Most importantly, this induction requires a wild-type azhA gene. These results suggest that upon AZC feeding and only in the presence of AzhA, GABA is produced and induces the expression of gatA, strongly indicating that AZC is metabolized through the GABA pathway via the action of the AzhA hydrolase.

Expression of gatA under AZC induction. Total RNA was extracted from hyphae of WT, azhAΔ, ngnAΔ and azhAΔ ngnAΔ strains on appropriately supplemented MM incubated at 37 °C. (A) Representative image of RT-PCR-derived mRNA transcripts of gatA gene in non-inducible condition (–) and in the presence of β-alanine (β-ala) or AZC, and fructose (Fr) as carbon source. (B) Relative quantification and statistical analysis of gatA transcripts in non-induced condition (–), β-ala or AZC, normalized by 18S rRNA levels. Statistical significance was analyzed via two-way ANOVA and is depicted with asterisks (*). Single (*), double (**) or triple (***) asterisks, indicate 0.01 ≤ p < 0.05, 0.001 < P ≤ 0.01, or p < 0.001, respectively.
In total, our results show that AZC is not catabolized through the L-Proline metabolic pathway, and it rather requires an intact GABA pathway. Moreover, AZC-mediated induction of both ngnA and azhA genes suggest an AmdR/IntA- and GABA-independent mechanism of their regulation at the transcriptional level (see also discussion).
Heterologous expression of AzhA hydrolase in S. cerevisiae
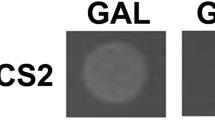
Τhe Σ1278b strain of S. cerevisiae containing the gene encoding the acetyltransferases Mpr1/2, orthologues of the NgnΑ acetyltransferase in A. nidulans, is resistant to AZC, but unable to utilize AZC as a nitrogen source19. Therefore, heterologous expression of AzhA in Σ1278b was used to confirm that AzhA hydrolase is necessary and sufficient for AZC-assimilation, since this strain is known to be able to use GABA as a nitrogen source48. Towards that, Σ1278b was transformed individually with a plasmid containing the azhA gene expressed under the gal1 promoter, which is activated in the presence of galactose49, and with an empty vector as a control (Tables 2 and 3). Our results show that only upon induction of AzhA expression (galactose as carbon source), Σ1278b strain becomes able to utilize AZC as a nitrogen source (Fig. 4A). It is additionally known that the widely used strain of S. cerevisiae, S288C, does not possess the Mpr1/2 acetyltransferases, and is therefore AZC-sensitive18. We thus used this strain to complement the lack of Mpr1/2 by heterologous expression of A. nidulans NgnΑ acetyltransferase (Fig. 4B). Using the approach described above, the S288C strain was transformed separately, with plasmids expressing ngnΑ and/or azhA genes, under the control of the gal1 promoter, and with an empty vector as a control (Tables 2 and 3). Our results are consistent with NgnA being an orthologue of Mpr1 and thus conferring the ability to the S288C strain to detoxify AZC (Fig. 4B).

Growth assays of S. cerevisiae strains expressing the A. nidulans azhA and ngnA ORFs. (A) Serial dilution assays of Σ1278b strains expressing either the azhA ORF under the regulation of the Gal1 glucose-repressible promoter, or the corresponding empty vector. The Σ1278b strain expressing the azhA ORF (azhA) is able to utilize AZC as a nitrogen source only in the presence of galactose (Gal) as a carbon source (AZC/Gal). (B) Serial dilution assays of S288C strains expressing either the A. nidulans the azhA and / or the ngnΑ ORFs under the regulation of the Gal1 promoter, or the corresponding empty vector. The S288C strain expressing either the azhA or the ngnΑ ORF is able to detoxify AZC.
In total, our results show that AzhA hydrolase is necessary and sufficient for the detoxification of AZC and its utilization as a nitrogen source in A. nidulans, most probably via the GABA assimilation pathway, while NgnΑ acetyltransferase additionally contributes to the detoxification of AZC.
Homologues of AzhA are scarcely found among bacterial and fungal species
To investigate whether proteins homologous to AzhA exist in species belonging to the three domains of life (Archaea, Bacteria and Eukaryotes) we searched the Database of National Center for Biotechnology Information (NCBI) with BLASTp, using the AzhA amino acid sequence, as query. The parameters of our searches were at least 80% coverage of the query sequence and E-values lower than 1e-05. Multiple sequence alignment indicated the conservation of the four highly conserved sequence motifs by which HAD-family members are recognized (in detail,—FDxDG,—T/SXx,—KPxP, and ED or GDxxxDD)28,47,48,52 among the fungal species that were retrieved by our BLASTp search (Supplementary Figure. S2) and were used for our phylogenetic analysis.
Moreover, sequence similarity to AzhA was found in all three major phylogenetic domains—i.e. Archaea, Bacteria and Eukaryotes (see Table S3 in the supplementary material). In eukaryotes, homologues of AzhA are mainly found in specific fungal taxa. Surprisingly enough, in the remaining eukaryotes, AzhA homologues were retrieved as single hits in species belonging to the lower eukaryotes (plakozoan, Trichoplax adhaerens) and to the kingdoms of Plantae (Quercus suber) and Metazoa (lepidopteron, Eumeta japonica).
Within the kingdom of Fungi AzhA, homologues are mainly found in species of the Eurotiales Order. On the contrary, both homologues of the amino or nucleic acid sequences of AzhA are missing from the genomes of the Saccharomycotina subphylum and the early diverging fungal lineages, such as Microsporidia, Chytridiomycota, Glomeromycota or Zygomycota53. Τhe only exception was in the subphylum of Mucomycotina, where only five hits were found. The amino acid matrix of AzhA homologues produced phylogenetic trees, which were almost invariant to the method applied (see Fig. S3 and S4 in the supplementary material).
All of our data were included in a “Universal tree”, in which, AzhA homologues are shown to split into two main clusters (Fig. 5). The first one consists of proteins mostly from Archaea, several Classes of Bacteria (i.e., Cyanobacteria, Delta-proteobacteria, Bacilli, Negativicutes, Erycipelotrichia, Clostridia, Spirochaetia and Halobacteria), and a few homologues from fungal classes which cannot be found in the second cluster (Dacrymycetales, Mixiales, Mucorales, Orbiliales, Taphrinales and Ustilaginales). The second cluster consists of Bacteria belonging to the classes of Alpha-, Beta- and Gamma- proteobacteria, Fungi from the phyla of Basidiomycota and Ascomycota (with the exception of species belonging to Cluster 1) and the remaining few representatives of Eukaryota (Fig. 5). In Ascomycota most results are found in the leotiomyceta clade, which are divided among the classes of Dothideomycetes, Eurotiomycetes and Sordariomycetes. Accordingly, in the phylum of Basidiomycota most results are found in the classes of Agaricomycetes and Tremellomycetes. The discrimination of these two clusters is strongly supported by both methods used (PP: 99% and NJ-bootstrap: 92.65%).

Phylogenetic relations of AzhA homologues in the three domains of life (Archaea, Bacteria and Eukaryotes), as emerged by the implementation of a MCMC tree, under the evolutionary model LG + G + F. All topologies produced are in agreement with the NJ analysis used as an alternative method. The tree is divided into two big clusters, which are noted with a black circle on the node that discriminates them. The coloring of the highlighted species is as follows: blue, green, grey and pink for bacteria, plants, archaea and fungi, respectively. The Aciduliprofundum spp. was used to root the tree. Tree visualization was conducted with Figtree.
The A. nidulans AzhA protein examined in this work, is placed within the Eurotiales clade with excellent support (PP: 100% and NJ-bootstrap: 94.33%) as expected (see Fig. S3 and S4 in the supplementary material). Another interesting aspect of the topology of the tree is that the basal group of the second cluster contains Pseudomonas and other relative genera, all belonging to Gamma-proteobacteria.
In addition, we noticed the strong clustering (100% support with both statistical methods used) of the homologue belonging to the sole plant representative (Quercus suber) in the tree with the putative homologue sequence of Neofusicoccum parvum, an aggressive phytopathogen, member of the Botryosphaeriaceae51,52,56. The close clustering of these two species could possibly indicate a gene transfer event between a plant and a plant pathogen. Accordingly, our attention was drawn by the presence of a putative AzhA homologue in a Candidatus Colwellia aromaticivorans sp. (Fig. 5). Interestingly, it was recently reported that strains of Candidatus Colwellia aromaticivorans sp. can be used to degrade sea oil spills57. Overall, these sequence data could provide good evidence for the identification of horizontal gene transfer (HGT) events between plants and fungal phytopathogens, as it has been previously shown58.
Discussion
A. nidulans as a soil fungus59 could potentially share the same ecological niches with AZC producing plants. AZC possibly serves as an allelochemical among plants, fungi and bacteria, maintaining unknown balances among them, like several other plant secondary metabolites do60,61. In this work we showed that A. nidulans possesses a specific, dual mechanism in order to cope with AZC toxicity, but also to utilize it as a nitrogen source. This detoxification mechanism relies on the presence of a hydrolase, designated AzhA, and an auxiliary acetyltransferase, NgnA25. In the presence of AZC as a sole nitrogen source, azhAΔ ngnAΔ double deletion strain shows no residual growth in both solid and liquid media, reflecting the highly toxic effect of AZC, when intracellularly accumulating. Specifically, our microscopic observations showed an 80% inhibition of azhAΔ ngnΑΔ spore germination in the presence of AZC, a phenotype similar to that observed macroscopically in solid growth tests (see Fig. 1B; also see Supplementary Video S1 in the supplementary material, online).
Our phylogenetic analysis indicated the high conservation of AzhA sequence among several taxa of fungi and gamma-proteobacteria. Collectively, our results strongly suggest that AzhA is a true orthologue of the A2C Pseudomonas hydrolase22, and is responsible for AZC detoxification and assimilation in A. nidulans. Thus, it is highly probable that the AzhA enzyme of A. nidulans functions similarly to its Pseudomonas spp. homologue and converts AZC to a GABA-like product, which could subsequently be assimilated via the GABA utilization pathway. Indeed, our results indicate that utilization of AZC as a source of nitrogen is independent of the L-Pro catabolic pathway and rather requires an intact and functional GABA catabolic pathway. More specifically, we show that the presence of AZC induces the expression of the GABA-inducible GatA GABA transaminase, and this requires an intact azhA gene, strongly suggesting that AzhA hydrolyses AZC to a GABA-like intermediate. The latter would promote the induction of gatA expression. We also show that heterologous expression of azhA in different S. cerevisiae strains is sufficient for AZC detoxification. Since these yeast strains are known to be able to use GABA as a sole nitrogen source62, this result shows that the sole reason for the lack of AZC utilization in yeasts is the lack of an AZC hydrolase, and is yet another indication that AzhA converts AZC to a GABA-like intermediate. It is also noteworthy that, contrary to S. cerevisiae, AZC uptake in A. nidulans is not mediated via neither the major L-Pro or GABA transporters, PrnB or GabA respectively, nor via the product of AN5678, the closest homologue of the General amino-acid permease Gap137. More work is needed in order to identify the AZC transport systems of the fungus, whose expression and activity seems to be, at least partially, nitrogen metabolite repression-dependent and carbon catabolite repression-independent. Our results support the scenario that AZC itself acts as the inducer for the expression of both azhA and ngnA genes. More specifically, AZC is still able to induce the expression of ngnA in the azhA- strain that is unable to metabolize AZC (Fig. 2), showing that conversion of AZC to a GABA-like intermediate is not required for the induction of ngnA. In addition, ngnA and azhA expression does not depend on, AmdR/IntA, the specific transcriptional activator of the GABA catabolic pathway genes. In fact, AZC causes an over-induction of ngnΑ in the amdR- strain. Given that this strain is unable to metabolize GABA, the above suggest that intracellular overaccumulation of AZC or GABA leads to increased expression of the AZC-detoxifying enzymes. GABA supplementation, however, does not induce the expression of ngnΑ and azhA (Fig. 2), strongly indicating that the reason for ngnΑ over-expression in the amdR- strain should be AZC over-accumulation. Importantly, expression of ngnA does not directly depend on either AreA or CreA general transcription factors either, as evidenced by similar basal and AZC-dependent induced levels of ngnΑ expression in the wild-type strain under both nitrogen (NH4+) and carbon (glucose; Gl) repressed conditions of growth compared to derepressed conditions (urea and fructose, respectively). Similarly, azhA expression is AreA and CreA independent (Fig. 2A,B). Thus, transcriptional regulation of ngnΑ and azhA expression differs from the usual situation of enzymes involved in the assimilation of secondary carbon and nitrogen sources in A. nidulans, that are under nitrogen metabolite repression and carbon catabolite repression36,63,64. An interesting explanation for this difference would be that the mechanism of ngnΑ and azhA induction is evolutionarily adapted to confer protection from AZC toxicity under all conditions of nutrient availability, given that AZC import into the cells seems to be mediated by transport systems that are also independent of nitrogen metabolite repression and carbon catabolite repression (see above).
Interestingly, we found ngnA basal expression levels to be quite low in non-induced conditions, and a slow migrating band in the absence of AZC, most probably corresponding to unspliced mRNA, is observed. In the presence of AZC this band disappears, suggesting an AZC-dependent splicing of ngnA upon its AZC-mediated induction. This observation is reminiscent of a recent report in mammalian cells, where intron retention of several transcripts was shown to correlate with nuclear localization of the mRNA65. It would, thus, be tempting to speculate that the splicing and nuclear export of the ngnA mRNA could be promoted, via an unknown mechanism, by the presence of its own substrate.
Our work provides evidence that AzhA is a member of the HAD superfamily, which encompasses various enzymes that catalyze carbon or phosphoryl group transfer reactions on a diverse range of substrates, using an active site of aspartate in nucleophilic catalysis50. Members of this enzyme superfamily have been shown to participate in the detoxification of xenobiotics and several other toxic substances28, being in agreement with our results suggesting AzhA as a detoxifying enzyme. Moreover, recent reports have proposed that some members of this family are genetically associated with human diseases such as cancer, cardiovascular, metabolic and neurological disorders66. In this work we employed a sequence-based phylogenetic analysis in order to explore the evolutionary history of AzhA. As expected, AzhA is located into the group of homologues belonging to Eurotiales. However, this phylogenetic relation was restricted to the taxonomic level of the Order and in some cases to the level of Class. This result is in contrast with single gene phylogenies of housekeeping genes, like beta-tubulin67 or the mitochondrial rps368, where the evolution of the examined sequences coincides with the evolution of the organism carrying them. This is expected since single gene phylogenies do not represent the evolutionary history of the whole organism but only of the gene itself6970. Moreover, our phylogenetic results indicate that AzhA has homologues in several fungal, bacterial and archaeal species. We noticed the high sequence conservation of AzhA amongst certain taxa in the kingdom of fungi, like the genera of Aspergilli, Penicillia and the order of Agaricales. On the contrary, we also noticed the complete absence of homologues in several other taxa, like the subdivision of Saccharomycotina. The most plausible interpretation of these results would be the presence of an AzhA homologue before the division of Dikarya, probably in the last fungal common ancestor; and afterwards, a series of several horizontal gene transfers (HGT) and losses throughout its evolution, which led to the corresponding presence or absence to the several fungal taxa. The relative posed question would be how this ancestral AzhA homologue “reached” the last common fungal ancestor, was it a case of horizontal gene transfer or was an endosymbiotic gene transfer event (EGT)? The presence of putative AzhA homologues in several Archaea species could provide some theoretical indications for a probable EGT event, even before the genesis of the last fungal common ancestor.
The most widely accepted endosymbiotic theory of the LECA71, supports the notion that the nuclear genome of eukaryotes derives from an ancestral state of a chimeric Archaeon72,73. From our analysis, this is reflected in the case of AzhA from the clustering of the early diverging fungal homologues (i.e. from Mucoromycotina) and the few other fungal homologues with the Archaea (Fig. 5). The rest fungal homologues might have been lost in the early evolutionary years in the different fungal taxa, like in the case of Saccharomycotina, and regained through independent HGT events from bacterial homologues (Fig. 5). The reasoning for reacquiring this gene and in extent this protein could be the evolutionary advantage it offers in ecological niches, where AZC could provide for nitrogen or carbon source. Some of these niches are common between fungi and bacteria, since the majority of these fungal and bacterial species are soil residents. This association of habitat and gene gain or loss events, could be also a possible explanation for the absence of AzhA in Saccharomycotina, since these species are not residents of soil74.
Overall, in this work, we propose the existence of a number of AzhA ancestor homologue/s, as members of the HAD superfamily, which have been either transferred in several fungal, bacterial or archaeal species through HGT events, or, alternatively, the presence of an old homologue before the existence of the last common fungal ancestor, who might have been the mediator for the HGT events among the later species.
Finally, based on the presented data and in the absence of AzhA homologues in AZC-producing plants, we propose that AzhA hydrolase contributes to a defense mechanism developed by microorganisms coexisting with AZC-producing plants. Our phylogenetic analysis indicated the presence of AzhA in a) several human dermatophyta (such as Histoplasma capsulatum, Blastomyces dermatidis etc.), b) fungal phytopathogens of great agricultural importance (such as Ustilago maydis and Magnaporthe oryzae etc.) and c) in bacteria degrading toxic substances (Candidatus Colwellia aromaticivorans). In total, the identification of enzymes like AzhA and NgnA could provide the basis for the identification of new antifungal pharmaceutical compounds against important human and plant fungal pathogens. Additionally, AzhA could be used as a new positive selection marker for transformation in ΑΖC-sensitive yeast and plants75,76. Accordingly, as a member of the HAD family, AzhA could be tested for potential detoxifying properties upon other toxic compounds and could be used in synthetic biology, for the development of new detoxifying systems.
Materials and methods
Media and growth conditions
A. nidulans cells were cultured in appropriately supplemented minimal media (MM) or complete media (CM) as previously described77,78 (and Fungal Genetics Stock Center, http://www.fgsc.net/) and grown at 25 and/or 37 °C. More precisely, nitrogen sources, ammonium tartrate (NH+4), urea (U), proline (Pro), GABA and AZC were used at a final concentration of 5 mM. Carbon sources, D-glucose (Gl), D-fructose (Fr) and ethanol (EtOH) were used at a final concentration of 1%, 0.1% and 3% (w/v), respectively. β-alanine was used at a final concentration of 50 mM (see RNA isolation and RT-PCR section).
S. cerevisiae cells were grown at 30 °C on a minimal buffered medium, pH 6.179, supplemented with galactose (Gal, 2% (w/v)) or glucose (Gl, 2% (w/v)) as carbon sources. Nitrogen sources, ammonium in the form of (NH4)2SO4, urea (U) or AZC were used at a final concentration of 5 mM. In S. cerevisiae the A. nidulans azhA and ngnA genes were under the control of the regulatable promoter gal1, which is repressed by glucose80,81.
Strains and Plasmids
A. nidulans strains and plasmids
The A. nidulans strains used in this study are listed in Table 1. Standard techniques for genetic analysis were used82. A. nidulans protoplast preparation and DNA transformation were performed as previously described83. For transformation of A. nidulans, DNA was isolated using the High Purity Plasmid kit (Roche), according to the manufacturer’s instructions and genomic DNA was prepared as described by Lee and Taylor (1990)84. The entire open reading frame of the azhΑ gene (ORF of AN12472) 726 bp long, was replaced in a recipient strain by the Aspergillus fumigatus pyrG gene (Afpyrg), using the fusion PCR gene replacement method and standard molecular cloning techniques85. The recipient strain carries a deletion of the nkuA gene (nkuAΔ), which is responsible for non-homologous end joining of DNA fragments. Deletion of the nkuA gene decreases the frequency of non-homologous integration of transforming DNA sequences into the A. nidulans genome, leading to improved gene targeting up to 90%83. The ∼1500 bp long, 5′ and 3′ flanking sequences of the azhA gene were PCR amplified using the Kapa-HiFi (Kapa Biosystems) DNA polymerase and primers 1–2 and 3–4, respectively (Supplementary Table S1). PCR products were digested with NotI/XbaI and XbaI/KpnI restriction endonucleases and subsequently cloned into a KpnI/NotI linearized pBlueScript II SK(+) vector (Table 3). In the unique XbaI site of the resulting plasmid, the AfpyrG gene was inserted, following its PCR amplification using primers 5 and 6 (Supplementary Table S1) from plasmid p143985,86 and XbaI digestion. The resulting cassette was PCR amplified using primers 1 and 4 and ∼2 μg of it, were used to transform the recipient strain. Selection of the transformants was carried out on MM with urea as sole nitrogen source. The intact, single copy, in-locus replacements were confirmed by Southern blot analysis using a non-radioactive, enzymatic DIG-labeled probe, prepared using the DIG Nonradioactive Nucleic Acid Labeling and Detection System (Roche), according to the manufacturer’s instructions. The azhAΔ ngnΑΔ double deletion strain was constructed by A. nidulans standard genetic crossing techniques87.
S. cerevisiae strains and plasmids
The S. cerevisiae strains used are listed in Table 2. They were constructed using the Σ1278b wild-type88 and BY4741 strains. Shuttle vectors, listed in Table 3, were used for the heterologous expression of A. nidulans azhA and ngnΑ genes into Σ1278b and BY4741 strains. Plasmids isolated were constructed by in vivo recombination in S. cerevisiae strains between linearized plasmids and PCR DNA fragments, using the primers reported in Supplementary Table S289. Each plasmid was isolated from transformed E. coli cells and confirmed by sequencing.
Microscopic observation of growth rate
Time lapse confocal microscopy was used for growth rate observations. Spores from freshly inoculated CM were collected and each spore suspension was used to inoculate supplemented liquid MM in µ-Slide 8 Well Chamber Slide (Ibidi GmbH, Germany). Cells were incubated for 18 h at 25 °C and observed by the use of a Leica TCS SP8 (Leica Microsystems Ltd., Milton Keynes, UK) confocal microscope. Growing hyphal tips were recorded with brightfield microscopy at high magnification, using a Leica HC PLΑΝ APO 63x/1.40NA oil immersion objective, and digital images captured every 15 min for a total of 45 min. Following, registration was performed automatically using the ImageJ plug-in, Linear Stack Alignment with SIFT, and tip growth rates were calculated using the MTrackJ manual tracking plug-in90 in Fiji image processing package91. At least 40 cells were counted for every condition tested at each experiment. More precisely, the total number of cells (n) observed for each condition tested was: WT: Urea, n = 161; Urea + AZC, n = 168; AZC, n = 171, azhAΔ: Urea, n = 120; Urea + AZC, n = 178; AZC, n = 126, ngn2Δ: Urea, n = 301; Urea + AZC, n = 202; AZC, n = 273, azhAΔ ngn2Δ: Urea, n = 271; Urea + AZC, n = 318; AZC, n = 362).
RNA isolation and RT-PCR
For total RNA extraction, A. nidulans conidiospores from ¼ of a 4-day CM plate were suspended in MM, filtered through blutex, and used to inoculate appropriately supplemented liquid MM. Mycelia were grown with shaking and subsequently harvested as previously described92. Where indicated, carbon and nitrogen sources were added as follows: D-glucose at a final concentration of 1%, D-fructose at 0.1%, EtOH at 3%, urea at 5 mM and ammonium, as tartrate salt at 5 mM. For induction, L-Pro, GABA or AZC were added at a final concentration of 20 mM and β-alanine at 50 mM. Mycelia were incubated at 25 or 37 °C for a total of 16 h. In order to examine the gatA gene expression, mycelia were grown at 37 °C for 26 h on MM with fructose and ammonium tartrate as carbon and nitrogen sources, respectively, as previously described by Richardson et al. (1989)27. Following, mycelia were thoroughly washed with ice-cold MM and about 1 g (net weight) was transferred aseptically in MM with fructose as carbon source, and 50 mM β-alanine or 20 mM AZC as nitrogen sources, respectively and incubated for additional 4 h.
RNA samples were prepared as previously descripted92,93, using the TRI Reagent (Sigma-Aldrich) kit, according to manufacturer’s instructions. Moreover, to avoid contamination with genomic DNA, 10 μg of each RNA sample were treated and cleaned up with TURBO DNA-free kit (Invitrogen). The absence of DNA contamination was verified by a conventional PCR (approximately 30 cycles), using specific- azhA, ngnA, gatA and 18S rRNA primers (Supplementary Table S1, primers 7 to 14,) and at least 100 ng of each RNA sample as template. The quality of isolated RNAs was examined by conventional gel electrophoresis using a 2% agarose gel stained with ethidium bromide (0,5 μg/ml). The concentration of each RNA sample was calculated using Nanodrop equipment (ND-1000 Spectrophotometer) according to the instructions of the manufacturer.
Approximately 100 ng, of each RNA sample, were used for reverse transcription PCR (RT-PCR) using the PrimeScript RT master mix (Perfect Real Time) (Takara Bio), according to manufacturer’s instructions. Primers for RT-PCR were designed so that the amplification fragment includes sequences belonging to at least one intron of each gene tested (except for azhA which is an intron-less gene) and were expected to produce amplicons of 355 bp (azhA cDNA), 478 bp (ngnA cDNA), 371 bp (gatA cDNA) and 280 bp (18S ribosomal RNA gene) (Supplementary Table S1). Densitometry analysis of RT products on an EtBr-stained 1% agarose gel was performed using ImageJ 1.43 (NIH) software. Relative optical density was calculated by dividing the densitometry of gene-specific cDNA with the respective loading control (18S rRNA).
Statistical analysis
Data were presented as mean ± SEM. GraphPad Prism software, version 8.0 was used for all statistical analyses. Two-way ANOVA with the nonparametric Kruskal–Wallis test, and Dunn’s multiple-comparison post hoc analyses were used to assess the significance of the value differences of all measurements.
Data collection
The Basic Local Alignment Search Tool for proteins (BLASTp) was used to retrieve amino acid sequences similar to AzhA, across the three main phylogenetic Domains (Archaea, Bacteria and Eukaryotes). AzhΑ hydrolase amino acid sequence was used as the query sequence throughout the NCBI database (retrieved on September 2019). The parameters used for sequence selection were: E-value threshold 10–05 maximum, query coverage 75% minimum and percentage identity at least 25%, using the default algorithmic settings to run the blast. In classes, where, too many sequences were retrieved, sequences of maximum homology according to the mentioned criteria were selected by all the representative orders.
Multiple sequence alignment
The collected sequences were aligned using the Clustal Omega (https://www.ebi.ac.uk/Tools/msa/clustalo)94 and the Jalview Version 2 programs95. Alignment parameters were set to default and results were inspected and manually edited in the sequence editor BioEdit, when necessary (http://www.mbio. ncsu.edu/BioEdit/bioedit.html, last accessed September 2019).
Phylogenetic analysis
Molecular evolutionary analyses were conducted by Neighbor Joining analysis (NJ), using the PAUP program96 and by Bayesian analysis, using the MrBayes program95,96,99. Reliability of nodes was assessed using 10 M bootstrap iterations for the NJ analyses. For the Bayesian analysis, the model of protein evolution that best fits the AzhA amino acid multiple sequence alignment was selected using the ProtTest program100. For the matrix containing all the fungal sequences the best fitting model (LG + I + G + F; alpha = 1) was determined according to BIC (Bayesian Information Criterion) and DT (decision theory). For the matrix containing AzhA similar sequences from all organisms, the best fitting model (LG + I + G + F; alpha = 1.47) was determined according to AIC (Akaike Information Criterion). The ProtTest program was also used for the estimation of the proportion of invariable sites and the alpha parameter of the gamma-distributed substitution rates. In all matrices (i.e. the matrices from all fungal strains and from all organisms), four independent MCMC searches were performed for each data set employing different random starting points (number of generations = 5,000,000), with sampling every 5000 generations. Convergence was inspected visually by plotting likelihood scores versus generation for the two runs. Based on this analysis, the burn-in was set to 10,000 in all cases. The program FigTree was used for the visualization of the trees and to make changes for their better presentation101 and phylo.io102 was used for the comparison of the trees constructed by the two different methods (data upon request). In all produced cases an automated comparison was conducted with plylo.io and the trees were found similar and most importantly identical at the topologies analyzed in the Results section.
References
Rubenstein, E., Zhou, H., Krasinska, K. M., Chien, A. & Becker, C. H. Azetidine-2-carboxylic acid in garden beets (Beta vulgaris). Phytochemistry 67, 898–903 (2006).
Rosenthal, G. Plant Nonprotein Amino and Imino Acids : Biological, Biochemical and Toxicological Properties (Elsevier, New York, 1982).
Fowden, L. Azetidine-2-carboxylic acid: a new constituent of plants. Nature 176, 347–348 (1955).
Fraenkel, G. S. The Raison d’Etre substances of secondary plant. Science 129, 1466–1470 (1959).
Stamp, N. Out of the quagmire of plant defense hypotheses. Q. Rev. Biol. 78, 23–55 (2003).
Samardzic, K. & Rodgers, K. J. Cell death and mitochondrial dysfunction induced by the dietary non-proteinogenic amino acid l-azetidine-2-carboxylic acid (Aze). Amino Acids 51, 1221–1232 (2019).
Fowden, L. Azetidine-2-carboxylic acid: a new cyclic imino acid occurring in plants. J. Ind. Microbiol. Biotechnol. 64, 323–332 (1956).
Fowden, L. & Richmond, M. H. Replacement of proline by azetidine-2-carboxylic acid during biosynthesis of protein. Biochim. Biophys. Acta 71, 459–461 (1963).
Song, Y. et al. Double mimicry evades tRNA synthetase editing by toxic vegetable-sourced non-proteinogenic amino acid. Nat. Commun. 8, 1–8 (2017).
Rodgers, K. J. & Shiozawa, N. Misincorporation of amino acid analogues into proteins by biosynthesis. Int. J. Biochem. Cell Biol. 40, 1452–1466 (2008).
Nomura, M., Nakamori, S. & Takagi, H. Characterization of novel acetyltransferases found in budding and fission yeasts that detoxify a proline analogue, azetidine-2-carboxylic acid. J. Biochem. 133, 67–74 (2003).
Lane, J. M., Parkes, L. J. & Prockop, D. J. Effect of the proline analogue azetidine-2-carboxylic acid on collagen synthesis in vivo II. Morphological and physical properties of collagen containing the analogue. Biochim. Biophys. Acta Protein Struct. 236, 528–541 (1971).
Zagari, A., Némethy, G. & Scheraga, H. A. The effect of the L -azetidine-2-carboxylic acid residue on protein conformation. I. Conformations of the residue and of dipeptides. Biopolymers 30, 951–959 (1990).
Trotter, E. W., Berenfeld, L., Krause, S., Petsko, G. & Gray, J. V. Protein misfolding and temperature up-shift cause G1 arrest via a common mechanism dependent on heat shock factor in Saccharomycescerevisiae. Proc. Natl. Acad. Sci. U. S. A. 98, 7313–7318 (2001).
Bach, T. M. H. & Takagi, H. Properties, metabolisms, and applications of l-proline analogues. Appl. Microbiol. Biotechnol. 97, 6623–6634 (2013).
Peterson, P. J. & Fowden, L. Different specificities of proline-activating enzymes from some plant species. Nature 200, 148–151 (1963).
Takagi, H., Iwamoto, F. & Nakamori, S. Isolation of freeze-tolerant laboratory strains of Saccharomyces cerevisiae from proline-analogue-resistant mutants. Appl. Microbiol. Biotechnol. 47, 405–411 (1997).
Takagi, H., Shichiri, M., Takemura, M., Mohri, M. & Nakamori, S. Saccharomyces cerevisiae Σ1278b has novel genes of the N-acetyltransferase gene superfamily required for L-proline analogue resistance. J. Bacteriol. 182, 4249–4256 (2000).
Shichiri, M., Hoshikawa, C., Nakamori, S. & Takagi, H. A novel acetyltransferase found in Saccharomyces cerevisiae Σ1278b that detoxifies a proline analogue, azetidine-2-carboxylic acid. J. Biol. Chem. 276, 41998–42002 (2001).
Kimura, Y., Nakamori, S. & Takagi, H. Polymorphism of the MPR1 gene required for toxic proline analogue resistance in the saccharomyces cerevisiae complex species. Yeast 19, 1437–1445 (2002).
Wada, M. et al. Distribution of L-azetidine-2-carboxylate N-acetyltransferase in yeast. Biosci. Biotechnol. Biochem. 72, 582–586 (2008).
Gross, C., Felsheim, R. & Wackett, L. P. Genes and enzymes of azetidine-2-carboxylate metabolism: detoxification and assimilation of an antibiotic. J. Bacteriol. 190, 4859–4864 (2008).
Andréasson, C., Neve, E. P. A. A. & Ljungdahl, P. O. Four permeases import proline and the toxic proline analogue azetidine-2-carboxylate into yeast. Yeast 21, 193–199 (2004).
Nützmann, H. W. et al. Bacteria-induced natural product formation in the fungus Aspergillus nidulans requires Saga/Ada-mediated histone acetylation. Proc. Natl. Acad. Sci. USA 108, 14282–14287 (2011).
Gournas, C., Evangelidis, T., Athanasopoulos, A., Mikros, E. & Sophianopoulou, V. The Aspergillus nidulans proline permease as a model for understanding the factors determining substrate binding and specificity of fungal amino acid transporters. J. Biol. Chem. 290, 6141–6155 (2015).
Andrianopoulos, A. & Hynes, M. J. Cloning and analysis of the positively acting regulatory gene amdR from Aspergillus nidulans. Mol. Cell. Biol. 8, 3532–3541 (1988).
Richardson, I. B., Hurley, S. K. & Hynes, M. J. Cloning and molecular characterisation of the amdR controlled gatA gene of Aspergillus nidulans. Mol. Gen. Genet. 217, 118–125 (1989).
Koonin, E. V. & Tatusov, R. L. Computer analysis of bacterial haloacid dehalogenases defines a large superfamily of hydrolases with diverse specificity. J. Mol. Biol. 244, 125–132 (1994).
Trinci, A. P. J. A kinetic study of the growth of Aspergillus nidulans and Other Fungi. J. Gen. Microbiol. 57, 11–24 (1969).
Skromne, I., Sanchez, O. & Aguirre, J. Starvation stress modulates the expression of the Aspergillus nidulans brlA regulatory gene. Microbiology 141, 21–28 (1995).
Tazebay, U. H., Sophianopoulou, V., Cubero, B., Scazzocchio, C. & Diallinas, G. Post-transcriptional control and kinetic characterization of proline transport in germinating conidiospores of Aspergillus nidulans. FEMS Microbiol. Lett. 132, 27–37 (1995).
Tazebay, U. H., Sophianopoulou, V., Scazzocchio, C. & Diallinas, G. The gene encoding the major proline transporter of Aspergillus nidulans is upregulated during conidiospore germination and in response to proline induction and amino acid starvation. Mol. Microbiol. 24, 105–117 (1997).
Hutchings, H. et al. The multiply-regulated gabA gene encoding the GABA permease of Aspergillus nidulans: A score of exons. Mol. Microbiol. 32, 557–568 (1999).
Arst, H. N. Integrator gene in Aspergillus nidulans. Nature 262, 319–324 (1976).
Sophianopoulou, V., Suárez, T., Diallinas, G. & Scazzocchio, C. Operator derepressed mutations in the proline utilisation gene cluster of Aspergillus nidulans. MGG Mol. Gen. Genet. 236, 209–213 (1993).
Arst, H. N. & Cove, D. J. Nitrogen metabolite repression in Aspergillus nidulans. MGG Mol. Gen. Genet. 126, 111–141 (1973).
Jauniaux, J.-C. & Grenson, M. GAP1, the general amino acid permease gene of Saccharomyces cerevisiae Nucleotide sequence, protein similarity with the other bakers yeast amino acid permeases, and nitrogen catabolite repression. Eur. J. Biochem. 190, 39–44 (1990).
Nishimura, A., Kotani, T., Sasano, Y. & Takagi, H. An antioxidative mechanism mediated by the yeast N-acetyltransferase Mpr1: oxidative stress-induced arginine synthesis and its physiological role. FEMS Yeast Res. 10, 687–698 (2010).
Tudzynski, B. Nitrogen regulation of fungal secondary metabolism in fungi. Front. Microbiol. 5, 1–15 (2014).
Sophianopoulou, V. & Diallinas, G. Amino acid transporters of lower eukaryotes: regulation, structure and topogenesis. FEMS Microbiol. Rev. 16, 53–75 (1995).
Cazelle, B., Pokorska, A., Hull, E., Green, P. M., Stanway, G., & Scazzocchio, C. Sequence, exon-intron organization, transcription and mutational analysis of prnA, the gene encoding the transcriptional activator of the prn gene cluster in Aspergillus nidulans. Mol. Microbiol. 28(2), 355–370 (1998).
Al Taho, N. M., Sealy-Lewis, H. M. & Scazzocchio, C. Suppressible alleles in a wide domain regulatory gene in Aspergillus nidulans. Curr. Genet. 8, 245–251 (1984).
Wilson, R. A. & Arst, H. N. Mutational analysis of AREA, a transcriptional activator mediating nitrogen metabolite repression in Aspergillus nidulans and a member of the “Streetwise” GATA family of transcription factors. Microbiol. Mol. Biol. Rev. 62, 586–596 (1998).
Arst, H. N., Tollervey, D., Dowzer, C. E. A. & Kelly, J. M. Notes an inversion truncating the creA gene of Aspergillus niduians results in carbon catabolite derepression. Mol. Microbiol. 4, 851–854 (1990).
Veen, P. Van Der, Ruijter, G. J. G. & Visser, J. An extreme creA mutation in Aspergillus nidulans has severe effects on D-glucose utilization. 2301–2306 (1995).
Katz, M. E. & Hynes, M. J. Characterization of the Amd R-controlled lama and lamb genes of Aspergillus-Nidulans. Genetics 122, 331–339 (1989).
Hynes, M. J. & Pateman, J. A. J. The genetic analysis of regulation of amidase synthesis in Aspergillus nidulans. II. Mutants resistant to Fluoroacetamide. MGG Mol. Gen. Genet. 108, 107–116 (1970).
André, B., Hein, C., Grenson, M. & Jauniaux, J. C. Cloning and expression of the UGA4 gene coding for the inducible GABA-specific transport protein of Saccharomyces cerevisiae. MGG Mol. Gen. Genet. 237, 17–25 (1993).
Flick, J. S. & Johnston, M. Two systems of glucose repression of the GAL1 promoter in Saccharomyces cerevisiae. Mol. Cell. Biol. 10, 4757–4769 (1990).
Burroughs, A. M., Allen, K. N., Dunaway-Mariano, D. & Aravind, L. Evolutionary genomics of the HAD superfamily: understanding the structural adaptations and catalytic diversity in a superfamily of phosphoesterases and allied enzymes. J. Mol. Biol. 361, 1003–1034 (2006).
Caparrós-Martín, J. A., McCarthy-Suárez, I. & Culiáñez-Macià, F. A. HAD hydrolase function unveiled by substrate screening: enzymatic characterization of Arabidopsis thaliana subclass I phosphosugar phosphatase AtSgpp. Planta 237, 943–954 (2013).
Toyoda, M. et al. Crystallization and preliminary X-ray analysis of L-azetidine-2-carboxylate hydrolase from Pseudomonas sp. strain A2C. Acta Crystallogr. Sect. F. Struct. Biol. Cryst. Commun. 66, 801–804 (2010).
James, T. Y. et al. Reconstructing the early evolution of Fungi using a six-gene phylogeny. Nature 443, 818–822 (2006).
Úrbez-Torres, J. R. & Gubler, W. D. Pathogenicity of Botryosphaeriaceae species isolated from grapevine cankers in California. Plant Dis. 93, 584–592 (2009).
Blanco-Ulate, B., Rolshausen, P. & Cantu, D. Draft genome sequence of Neofusicoccum parvum isolate UCR-NP2, a fungal vascular pathogen associated with grapevine cankers. Genome Announc. 1, e00339 (2013).
Iturritxa, E., Slippers, B., Mesanza, N. & Wingfield, M. J. First report of Neofusicoccum parvum causing canker and die-back of Eucalyptus in Spain. Australas. Plant Dis. Notes 6, 57–59 (2011).
Campeão, M. E. et al. “Candidatus Colwellia aromaticivorans” sp. nov., “Candidatus Halocyntiibacter alkanivorans” sp. nov., and “Candidatus Ulvibacter alkanivorans” sp. nov. genome sequences. Microbiol. Resour. Announc. 8, 15 (2019).
Nikolaidis, N., Doran, N. & Cosgrove, D. J. Plant expansins in bacteria and fungi: evolution by horizontal gene transfer and independent domain fusion. Mol. Biol. Evol. 31, 376–386 (2014).
Watkinson, S., Boddy, L. & Money, N. The Fungi (Academic Press, 2015). https://doi.org/10.1007/978-1-4684-3495-8_15.
Dunlop, R. A., Main, B. J. & Rodgers, K. J. The deleterious effects of non-protein amino acids from desert plants on human and animal health. J. Arid Environ. 112, 152–158 (2015).
Bertin, C. et al. Grass roots chemistry: meta-tyrosine, an herbicidal nonprotein amino acid. Proc. Natl. Acad. Sci. U. S. A. 104, 16964–16969 (2007).
McKelvey, J., Rai, R. & Cooper, T. G. Gaba transport in Saccharomyces cerevisiae. Yeast 6, 263–270 (1990).
Marzluf, G. A. Genetic regulation of nitrogen metabolism in the fungi. Microbiol. Mol. Biol. Rev. 61, 17–32 (1997).
Bailey, C. & Arst, H. N. Jr. Carbon catabolite repression in Aspergillus nidulans. Eur. J. Biochem. 51, 573–577 (1975).
Zeng, C. & Hamada, M. RNA-Seq analysis reveals localization-associated alternative splicing across 13 cell lines. Genes (Basel) 11, 820 (2020).
Seifried, A., Schultz, J. & Gohla, A. Human HAD phosphatases: structure, mechanism, and roles in health and disease. FEBS J. 280, 549–571 (2013).
Keeling, P. J. & Doolittle, W. F. Alpha-tubulin from early-diverging eukaryotic lineages and the evolution of the tubulin family. Mol. Biol. Evol. 13, 1297–1305 (1996).
Korovesi, A. G., Ntertilis, M. & Kouvelis, V. N. Mt-rps3 is an ancient gene which provides insight into the evolution of fungal mitochondrial genomes. Mol. Phylogenet. Evol. 127, 74–86 (2018).
Kouvelis, V. N., Ghikas, D. V. & Typas, M. A. The analysis of the complete mitochondrial genome of Lecanicillium muscarium (synonym Verticillium lecanii) suggests a minimum common gene organization in mtDNAs of Sordariomycetes: phylogenetic implications. Fungal Genet. Biol. 41, 930–940 (2004).
Pantou, M. P., Kouvelis, V. N. & Typas, M. A. The complete mitochondrial genome of Fusarium oxysporum: insights into fungal mitochondrial evolution. Gene 419, 7–15 (2008).
O’Malley, M. A., Leger, M. M., Wideman, J. G. & Ruiz-Trillo, I. Concepts of the last eukaryotic common ancestor. Nat. Ecol. Evol. 3, 338–344 (2019).
Bock, R. Witnessing genome evolution: experimental reconstruction of endosymbiotic and horizontal gene transfer. Annu. Rev. Genet. 51, 1–22 (2017).
Pittis, A. A. & Gabaldón, T. Late acquisition of mitochondria by a host with chimaeric prokaryotic ancestry. Nature 531, 101–104 (2016).
Jouhten, P., Ponomarova, O., Gonzalez, R. & Patil, K. R. Saccharomyces cerevisiae metabolism in ecological context. FEMS Yeast Res. 16, fow080 (2016).
Zhang, X. H., Takagi, H. & Widholm, J. M. Expression of a novel yeast gene that detoxifies the proline analog azetidine-2-carboxylate confers resistance during tobacco seed germination, callus and shoot formation. Plant Cell Rep. 22, 615–622 (2004).
Tsai, F. Y., Zhang, X. H., Ulanov, A. & Widholm, J. M. The application of the yeast N-acetyltransferase MPR1 gene and the proline analogue L-azetidine-2-carboxylic acid as a selectable marker system for plant transformation. J. Exp. Bot. 61, 2561–2573 (2010).
Pontecorvo, G., Roper, J. A., Chemmons, L. M., Macdonald, K. D. & Bufton, A. W. J. The genetics of Aspergillus nidulans. Adv. Genet. 5, 141–238 (1953).
Cove, D. J. The induction and repression of nitrate reductase in the fungus Aspergillus nidulans. Biochim. Biophys. Acta Enzymol. Biol. Oxid. 113, 51–56 (1966).
Jacobs, P., Jauniaux, J. C. & Grenson, M. A cis-dominant regulatory mutation linked to the argB-argC gene cluster in Saccharomyces cerevisiae. J. Mol. Biol. 139, 691–704 (1980).
Flick, J. S. & Johnston, M. Two systems of glucose repression of the GAL] promoter in Saccharomyces cerevisiae. Mol. Cell. Biol. 10, 4757–4769 (1990).
Johnston, M. A model fungal gene regulatory mechanism: the GAL genes of Saccharomyces cerevisiae. Microbiol. Mol. Biol. Rev. 51, 458–476 (1987).
Clutterbuck, A. J. Aspergillus nidulans. In Bacteria, Bacteriophages, and Fungi 447–510 (Springer, 1974). https://doi.org/10.1007/978-1-4899-1710-2_26
Nayak, T. et al. A versatile and efficient gene-targeting system for Aspergillus nidulans. Genetics 172, 1557–1566 (2006).
Lee, S. B. & Taylor, J. W. Isolation of DNA from fungal mycelia and single spores. In PCR Protocols 282–287 (Elsevier, 1990). https://doi.org/10.1016/b978-0-12-372180-8.50038-x
Szewcyk, E. et al. Fusion PCR and gene targeting in Asperigillus nidulans. Nat. Protoc. 1, 3111–3120 (2006).
Vangelatos, I. et al. Eisosome organization in the filamentous ascomycete Aspergillus nidulans. Eukaryot. Cell 9, 1441–1454 (2010).
Todd, R. B., Davis, M. A. & Hynes, M. J. Genetic manipulation of Aspergillus nidulans: heterokaryons and diploids for dominance, complementation and haploidization analyses. Nat. Protoc. 2, 822–830 (2007).
Grenson, M. Multiplicity of the amino acid permeases in Saccharomyces cerevisiae. Biochim. Biophys. Acta Gen. Subj. 127, 339–346 (1966).
Merhi, A. & Andre, B. Internal amino acids promote Gap1 Permease ubiquitylation via TORC1/Npr1/14-3-3-dependent control of the Bul Arrestin-like adaptors. Mol. Cell. Biol. 32, 4510–4522 (2012).
Meijering, E., Dzyubachyk, O. & Smal, I. Methods for cell and particle tracking. Methods Enzymol. 504, 183–200 (2012).
Schindelin, J. et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682 (2012).
Athanasopoulos, A., Boleti, H., Scazzocchio, C. & Sophianopoulou, V. Eisosome distribution and localization in the meiotic progeny of Aspergillus nidulans. Fungal Genet. Biol. 53, 84–96 (2013).
Athanasopoulos, A., Gournas, C., Amillis, S. & Sophianopoulou, V. Characterization of AnNce102 and its role in eisosome stability and sphingolipid biosynthesis. Sci. Rep. 5, 15200 (2015).
Thompson, J. D., Higgins, D. G. & Gibson, T. J. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22, 4673–4680 (1994).
Waterhouse, A. M., Procter, J. B., Martin, D. M. A., Clamp, M. & Barton, G. J. Jalview Version 2-A multiple sequence alignment editor and analysis workbench. Bioinformatics 25, 1189–1191 (2009).
L. Swofford, D. PAUP*. Phylogenetic Analysis Using Parsimony (*and Other Methods). Version 4.0b10. version 4.0b10 edn Version 4., (2002).
Mau, B., Newton, M. A. & Larget, B. Bayesian phylogenetic inference via Markov chain Monte Carlo methods. Biometrics 55, 1–12 (1999).
Huelsenbeck, J. P. & Ronquist, F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 17, 754–755 (2001).
Ronquist, F. & Huelsenbeck, J. P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19, 1572–1574 (2003).
Abascal, F., Zardoya, R. & Posada, D. ProtTest: selection of best-fit models of protein evolution. Bioinformatics 21, 2104–2105 (2005).
2009., R. A. FigTree. Available from: http://tree.bio.ed.ac.uk/software/figtree/.
Robinson, O., Dylus, D. & Dessimoz, C. Phylo.io: interactive viewing and comparison of large phylogenetic trees on the web. Mol. Biol. Evol. 33, 2163–2166 (2016).
Acknowledgements
We thank Prof. Bruno André for providing S. cerevisiae strains and plasmids. We are grateful to Dr. A. Fytrou for critically reading the manuscript. This work was partly supported by the projects “Target Identification and Development of Novel Approaches for Health and environmental Applications” (MIS 5002514) which is implemented under the Action for the Strategic Development on the Research and Technological sectors and “A Greek Research Infrastructure for Visualizing and Monitoring Fundamental Biological Processes (BioImaging-GR)” (MIS 5002755), which is implemented under the Action “Reinforcement of the Research and Innovation Infrastructure”. Both projects are funded by the Operational Programme “Competitiveness, Entrepreneurship and Innovation” (NSRF 2014-2020) and co-financed by Greece and the European Union (European Regional Development Fund).
Author information
Authors and Affiliations
Contributions
A.B. and V.S. designed the experiments. A.B. performed the experiments, analyzed and interpreted the data. A.A. discussed the results and provided critical assistance with microscopy experiments. C.G. provided assistance for yeast experiments and discussed the results. V.K. provided critical assistance for the phylogenetic analyses. V.S. Supervision, project administration and funding acquisition. Writing: A.B. original draft preparation. A.A. and C.G. and V.S. extensive Review and Editing.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Biratsi, A., Athanasopoulos, A., Kouvelis, V.N. et al. A highly conserved mechanism for the detoxification and assimilation of the toxic phytoproduct L-azetidine-2-carboxylic acid in Aspergillus nidulans. Sci Rep 11, 7391 (2021). https://doi.org/10.1038/s41598-021-86622-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-86622-3
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
