
Abstract
CNGCs are ligand-gated calcium signaling channels, which participate in important biological processes in eukaryotes. However, the CNGC gene family is not well-investigated in Brassica rapa L. (i.e., field mustard) that is economically important and evolutionary model crop. In this study, we systematically identified 29 member genes in BrCNGC gene family, and studied their physico-chemical properties. The BrCNGC family was classified into four major and two sub phylogenetic groups. These genes were randomly localized on nine chromosomes, and dispersed into three sub-genomes of B. rapa L. Both whole-genome triplication and gene duplication (i.e., segmental/tandem) events participated in the expansion of the BrCNGC family. Using in-silico bioinformatics approaches, we determined the gene structures, conserved motif compositions, protein interaction networks, and revealed that most BrCNGCs can be regulated by phosphorylation and microRNAs of diverse functionality. The differential expression patterns of BrCNGC genes in different plant tissues, and in response to different biotic, abiotic and hormonal stress types, suggest their strong role in plant growth, development and stress tolerance. Notably, BrCNGC-9, 27, 18 and 11 exhibited highest responses in terms of fold-changes against club-root pathogen Plasmodiophora brassicae, Pseudomonas syringae pv. maculicola, methyl-jasmonate, and trace elements. These results provide foundation for the selection of candidate BrCNGC genes for future breeding of field mustard.
Similar content being viewed by others


Introduction
CNGCs, i.e., Cyclic nucleotide-gated ion channels, are porous cation-conducting channels and elements of the signal transduction pathways that allows the transportation of calcium, sodium and potassium cations across the cell membranes1. Therefore, CNGC proteins are usually found within the cytoplasmic membrane2,3, vacuole membrane4, or nuclear membrane5. In animals, the CNGCs have been reported to transfer the signals required by sensory processes6. However, in plants, the CNGCs perform more diverse functions such as absorption of the essential and toxic cations, Ca2+ signalling, growth, fertility of pollen, geotropism, leaf senescence, inherent immunity, and tolerance to biotic and abiotic stress2,7,8,9,10. The CNGC-encoded proteins in animal system have been well characterized, but, research on plant CNGCs has just begun and these genes have been reported from limited plant species, comprising Arabidopsis thaliana1, rice11, pear12, tomato13, Physcomitrella patens14 and tobacco15. During our latest study of Brassica oleracea genome, we uncovered many hidden features of plant CNGCs including conserved domains, gene structures, phylogeny and evolution, function and underlying mechanisms16. For example, CNGC proteins have well-preserved domain structures comprising ion transport domain at N-terminus and cNMP-binding domain at C-terminus that embodies phosphate-binding cassette (PBC) and hinge region, thus regulates the closing and opening of channel via cAMP and/or cGMP11,17. Additionally, a Calmodulin (CaM) binding domain controls the activity of CNGC from inside the cell by calcium, CaM and cNMP which act as secondary messengers, and a conserved isoleucine–glutamine (IQ) at C-terminus motif that upon binding to the CaM regulates the channel activity2,17,18,19. Moreover, the plant CNGC families can be phylogenetically classified into four groups, and the member genes function in a group-dependent manner. For example, Group IV-a CNGCs are reported to function in salt stress tolerance4, while the Group IV-b genes may be involved disease and heat resistance20,21. It is not clear if this correspondingly applies to other plant species22.
Brassica rapa L. is one of the notable member of the Brassicaceae (also known as Cruciferae, mustard or the cabbage family) known for its agricultural and economic importance23,24. B. rapa L. is one such important vegetable plant with medicinal properties, and is highly consumed around the world25. Besides, the unique genome structure of B. rapa L, represented by multiple sub-genomic fractions and closed syntenic relation with B. oleracea and Arabidopsis, makes it an important model crop for studies involving plant genomics, evolution, breeding and molecular genetics. Dietary Brassica crops are important for their economic, nutritional (lutein, vitamin A, folate, vitamin C, vitamin K and calcium) and antioxidant properties26. It is believed that a high consumption of Brassica vegetables reduces the threat of age -related chronic diseases27 and lessens the risk of several types of cancer28,29. The genome of this important crucifer crop is sequenced and deposited as in Brassica database (BRAD). Taking advantage of the available genomic data, we performed genome-wide identification of the CNGC family in B. rapa L. We employed multiple in silico approaches to perform genomic and functional investigates of CNGC genes and proteins in field mustard, including systematic characterization, classification, phylogeny, synteny, evolution, and gene expression.
Results and discussion
Genome-wide identification of BrCNGC family
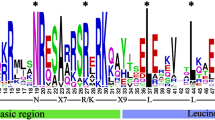
The CNGC genes play vital roles in development, ion transport, signaling and stress responses11,12,13, and the CNGC gene families have been studied in limited yet important crops30,31,32,33. However, the systematic identification and annotation of this family has not been performed in crucifer plants except for Chinese cabbage by our group recently16. The genomic sequence of B. rapa L., one of the most significant species of Brassica genus, was released in 201124. Therefore, proper annotation and identification of the CNGC genes in B. rapa L. was performed in this study. All non-redundant putative gene sequences were retrieved from BRAD database, and analysed for the presence of plant CNGC-specific conserved domains and motifs. Consequently, accessions either having truncated sequences or missing CNGC-specific domains were discarded from further analysis. For instance, accession Bra022235, was a short truncated sequence lacking essential plant CNGC-specific domains such as CNBD33. Finally, twenty-nine genes with full length amino acid sequences (> 500 aa) were identified and confirmed as members of the BrCNGC family (Table 1). Each protein of the BrCNGC family comprised a fully conserved CNBD and IT domains, with overlapped CaMBD and adjacent IQ domains (Fig. 1a,c). Within the CNBD, the two most conserved regions were identified: a PBC motif, which binds the sugar and phosphate moieties of the cNMP ligand, and a “hinge” region adjacent to the PBC, which is believed to contribute to ligand binding efficacy and selectivity17. Moreover, the latest criterion for identification of CNGC genes is the validation of CNGC-specific motif key, which upon failing can mislead both the readers and researchers regarding the plant CNGCs including their classification and overall structure as a family. Using multiple sequence alignment at > 90% conservation, we deduced the BrCNGC-specific consensus motif key [[L] – X (2)—[G] –X (3)-[G] –X (1,2)-L -L -X -W –X (0,1,2)-[L] –X (7,14)-[P] –X (1,5)-S-X (10)-[E] -X -[F] -X –L] (Fig. 1b). The key spanning the PBC and hinge region within the CNBD domain, recognizes all 29 BrCNGCs identified in this study.

Domain architecture, consensus motif key and conserved cNMP-binding domain (CNBD) alignment of BrCNGC family proteins. (a) Each protein of the BrCNGC family comprised a fully conserved CNBD and IT domains, with overlapped CaMBD and adjacent IQ domains. (b) Plant and Brassica-specific CNGC-recognizing specific consensus motif key deduced after multiple sequence alignment at > 90% conservation. (c) Multiple sequence alignment of BrCNGC proteins using CNBD domain.
Phylogenetic analysis and classification of BrCNGCs
It is anticipated that homologs belonging to the similar taxonomic clade probably also resemble in structural, functional and evolutionary properties34. Such information can be used in clarifying the role(s) of the newly identified BrCNGCs. The multiple sequence alignment using full length amino acid sequences and conserved domains showed > 90% resemblance of the representative BrCNGCs among themselves, and with their respective orthologs in A. thaliana (i.e., AtCNGCs) and B. oleracea (BoCNGCs) (Fig. 2; Supplementary Figs. S1–S4 and Tables S1−S2)16. Using neighbor-joining method, the BrCNGC gene family was classified into four main clades based on the classification of AtCNGCs, tree topology and bootstrap values (Fig. 2; Supplementary Fig. S4). The member BrCNGC genes were named based on their positions in phylogenetic tree. Among these, seven BrCNGC genes clustered in clade-I, five in clade-II, and six in clade-III. Clade-IV that was additionally separated into two sub-clade (IV-a and IV-b), contained highest number of BrCNGCs genes (i.e., 11). These findings were in covenant to the previous investigations1,15,16.

Phylogeny of CNGC proteins from Brassica rapa L. and A. thaliana. A maximum likelihood phylogenetic tree was created with MEGA 6.0, using the Jones–Taylor–Thornton model. The bootstrap values from 1000 replications are provided at each node. The BoCNGC proteins identified in this study are indicated with maroon diamonds, while the AtCNGCs are indicated with blue squares. Each group is highlighted in different color.
Chromosomal mapping and distribution on three sub-genomes
The 29 BrCNGC genes were unsystematically dispersed across the B. rapa L. genome and localized on nine of ten chromosomes (i.e., A01–07 and A09–A10). The distribution of BrCNGC genes on chromosomes was uneven, for example, chromosome A01 carried six genes, while others had 2–4 genes. None of BrCNGC genes was localized on chromosome A08. Among 29 BrCNGC genes, 15 loci were located on forward strand, while 14 loci were positioned on reverse strand of the chromosomes (Fig. 3). Similar to B. oleracea, the genome of B. rapa L. is currently fractioned into three sub-genomes: i.e., least fractionated (LF), medium fractionated (MF-I) and most fractionated (MF-II)24. The LF sub-genome of B. rapa L. contained maximum numbers of BrCNGC genes (i.e., 14 genes), while MF-II carried only 3 BrCNCG genes (Table 2). These findings are agreement to our previous findings of BoCNGC sub-genomes16.

Chromosomal localization and duplication of BrCNGC family genes. Physical location and distance of BrCNGC genes across the 9 chromosomes of B. rapa. BrCNGC genes are shown as numbers on chromosomes, tandemly duplicated gene pairs by white color, while segmental duplications are indicated with asterisks. Red and yellow lines show forward and reverse orientations of each loci respectively.
Evolution of BrCNGC family
Origin and comparative synteny analysis of BrCNGC family genes
B. rapa L. is an ancient polyploid, whose genome has undergone whole genome triplication (WGT) event ~ 13–17 million years ago (MYA), after divergence from A. thaliana, followed by large-scale re-diploidization (chromosomal re-arrangements)35. Being a member of the conventional triangle of U36, the assembled genome of B. rapa L. (312 Mb) is smaller than sister specie B. oleracea (540 Mb)37, which diverged from a common ancestor ~ 4 MYA38. Currently, the genomes of B. oleracea and B. rapa L. are categorised as LF, MFI and MF-II37. Because of a Brassica-lineage specific WGT, each A. thaliana CNGC gene was expected to generate three Brassica copies. However, there were 20 AtCNGC genes, 29 BrCNGC genes, and 26 BoCNGC genes. The LF, MF-I and MF-II sub-genomes, respectively retained 65%, 40% and 15% of the CNGC genes found in A. thaliana. To detect the retention or loss of CNGC genes after a WGT, the syntenic map of BrCNGC genes with the model A. thaliana and B. oleracea CNGC genes provided markers for defining the regions of conserved synteny among the three genomes (Supplementary Fig. S5) (Table 2). We found that more than > 80% of BrCNGC genes are located in well-conserved syntenic blocks, with deletion and gain of some genes, which coincides with the previous findings39. Compared with the ancestral Brassicaceae blocks (A to X) in A. thaliana, the synteny of 75% of the CNGC gene family was preserved in Brassica species, based on the number of corresponding genes. Ten of the 20 AtCNGC genes were retained as single copy in the equivalent blocks of both Brassica species. Three AtCNGC genes (i.e., AT2G23980, AT2G24610 and AT5G54250), located on I and W syntenic blocks, were preserved as two copies in Brassica genomes, which were asymmetrically fractionated into three sub-genomes. Two AtCNGC genes (i.e., AT3G17690 and AT3G17700) in F syntenic block were retained as three copies in each species. Two BrCNGC genes (i.e., BrCNGC1 and BrCNGC24) were respectively located on conserved syntenic block with BoCNGC3 and BoCNGC23, but not with AtCNGC genes. An extra gene copy (Bra022235) was located on potential overlap/tandemly repeated regions of F block along with gene pair BrCNGC26 and BrCNGC29 (Table 2). Thorough examination revealed that this gene has lost its functional CNBD domain during the course of evolution. These results are agreement to the findings of Duan et al.40 who reported that functionally redundant gene copies are lost after genome duplication event, while functionally important some gene copies are retained. Together, these finding suggest that WGT, along with segmental duplication played important role in expansion of BrCNGC gene family overall, while, tandem duplication was identified to play role in expansion of group IV-b only. Moreover, conservation of CNGC genes after substantial genome reshuffling event suggests that these genes are crucial for plant development41.
Gene duplication events and expansion of BrCNGC family
Gene family expands through one of three possible mechanisms including tandem and segmental duplication, and/or whole-genome duplication42. The examination of gene duplication events showed that three gene pairs (i.e., BrCNGC25/BrCNGC28, BrCNGC22/BrCNGC27 and BrCNGC26/BrCNGC29) are tandemly duplicated genes in B. rapa L. genome, as revealed by analysis in PTGBase. These tandemly duplicated genes are located on adjacent loci of chromosome 1, 3 and 5 respectively. In addition, 8 BrCNGC genes were likely associated with segmental duplications, which however require further elucidation (Fig. 3). These observations suggest that both tandem and segmental duplications may have donated to functional and enlargement diversity of BrCNGC gene family.
Gene structures and conserved motifs of BrCNGC-encoded proteins
The diversity in exon–intron play an imperative role in gene families evolution, which provide more evidences of phylogenetic clustering43. Here, we analyzed the exon–intron orderliness of the individual BrCNGC gene, and conserved motifs in their encoded protein sequences to describe the structural variety of the BrCNGC family. The most of the BrCNGC genes from phylogenetic clade I-III included six or seven exons, while, clade IV-b contained highest number of exons, ranging between 10 and 11 (Fig. 4). Nearly grouped BrCNGC genes in the similar clades were alike on the subject of the number of exons-introns sizes. Maximum of the introns in BrCNGC genes were phase-0 introns that exist in between complete codons. Thirty-three phase-1 introns that are separated by 1st codon and thirty-five phase-2 introns that are positioned in the middle of the second and third nucleotides of a codon were detected in the BrCNGC family. The exceptions were BrCNGC3, BrCNGC5 and BrCNGC9, which comprised three phase-1 introns. Comparison of exon–intron organization with the AtCNGC genes which clustered into similar phylogenetic groups shown numerous alterations (Supplementary Fig. S6). Utmost of the phase-1 and 2 introns were existing in AtCNGC genes, inferring that intron loss for the duration of evolution caused in a reduction in the number of introns in BoCNGC genes, principally those in clade I–III and IV-a (Supplementary Fig. S7).

Schematic diagram showing the structures of BrCNGC genes and conserved motifs in their encoded proteins. Exon–intron organization and conserved motifs within the 29 BrCNGCs. The NJ phylogenetic tree of CDs is shown on the left side of the figure, exons-introns indicated as blue boxes and red lines respectively, and motifs are represented by colored boxes within the exons. Numbers [0, 1 and 2] given on gene structures represent the respective intron phases. The lengths of each exon and intron can be mapped to the scale given in the bottom. The order of motifs corresponds to the position of the motifs in protein sequence, however, the length of the boxes does not correspond to the lengths of motifs.
Ten conserved motifs were identified in BrCNGCs during motif structure studies Multiple Expectation Maximization for Motif Elicitation suite (MEME)44. Rendering to Pfam codes45 and WebLogos, only six motifs (i.e., 1–4, 7, and 8) comprised domains with known functions (Fig. 4, Supplementary Fig. S8 and Table S3). Motif 1 was the biggest motif accompanying with product of unknown functions. Motifs 2, 3, 7, 8 and 4, which encode a CNBD, an ion transport domain, and IQ domain, correspondingly, were preserved among all BrCNGC family members. Notably, each clade members had similar arrangement of functionally annotated motifs, reveals that the directly associated proteins in each clade showed alike motif arrangements and perhaps functional resemblances too. The functionality of the leftover motifs (1, 5, 6, 9 and 10) wait for additional experimental evidence.
Protein sequence features and physico-chemical properties of BrCNGCs
The biochemical and physiological characteristics of the 29 BrCNGC proteins were identified (Table 3). The ProtParam tool showed that most of these proteins are localized in plasma-membrane. BrCNGC proteins varied in lengths from 556 to 786 aa with average of 711 aa, molecular weight (64.27–90.34 kDa), and residue weight (112.566–116.172 g/mol depending on the number of atoms present. Approximately, one-thirds of the BrCNGC proteins had low net charge (< 19) and relatively low isoelectric points (pI < 9). Approximately, all BrCNGC were hydrophilic, with BrCNGC21 and BrCNGC23 being somewhat hydrophobic. Based on the aliphatic index, most BrCNGC proteins were thermostable, similar to other globular proteins. Rendering to the instability index (II), none of the BrCNGC family proteins was stable in the test tube (Table 3). Additionally, the BrCNGC proteins had more positively charged residues than negatively charged residues (Supplementary Fig. S9). Hydrogen was the most abundant, followed by carbon, nitrogen and oxygen, and sulfur (Supplementary Fig. S10). Leucine was a very common amino acid among the 26 BrCNGC proteins (Supplementary Fig. S11).
Distribution of Post-translational modifications and microRNA target sites in BrCNGCs
Post-translational modifications (PTMs) of protein upturn the variety of their functions over and done with diverse mechanisms46. These mechanisms may include, protein localization, protein–protein interaction, cleavage, degradation or allosterically regulating enzyme activity47. We analysed BoCNGC protein sequences using ScanProsite48, multiple putative phosphorylation sites were identified (Table 4). These locations may act as substrates for numerous kinases, comprising tyrosine kinase, casein kinase II, cAMP/cGMP kinase, and protein kinase c. All proteins contained non-potential Glycosylphosphatidylinositol (GPI) anchor modification site in their sequences, while 16 BrCNGCs contained PEST-like sequences, which may act as a signal peptides for protein degradation49. Most abundant sites were casein kinase II sites, with 17 sites in BrCNGC7, followed by protein kinase C, were the maximum in clade IV members. All BrCNGC proteins had multiple N-glycosylation/ N-myristoylation motif locates are greatly preserved than rest of the PTMs. The rest of the PTM sites, such as those for amidations, leucine zipper patterns, and P-loop of the -GTP/ATP binding site motif A, were less preserved and arbitrarily dispersed, increasing diversity to function and mechanisms of CNGC-definite PTMs47. MicroRNAs (miRNAs) are interior non-coding RNAs that direct gene expression, particularly post-transcriptional gene silencing50. Recognising the targets of the expected miRNAs could facilitate the understandings of the genetic functions of miRNAs prompting signal transduction, stress adaptations, and plant development51. Herein, we investigated for possible miRNA targets in the set of recognized BrCNGC transcripts52. We recognized 92 miRNAs comprising target sites in 28 BrCNGC transcripts using a cut-off threshold of 5 for the search parameters (Supplementary Table S4). Small RNA/target site paired with an expectation score and cut-off threshold of 4 were included to reduce the number of false positive predictions. Consequently, seventeen miRNAs with target sites in fourteen BrCNGC genes were recognized, among which, four miRNAs with an expectation score < 3.5 can be considered more reliable (< 3.5) (Supplementary Table S5). Most of the BrCNGC genes included target site for single miRNA, except for BrCNGC14, BrCNGC20 and BrCNGC21, which contained target sites for 2 miRNAs. The convenience of the target site wide-ranging from 8.828 (bra-miR9552b-5p) to 20.9 (bra-miR160a-3p), where minor values resemble to a grander likelihood of interaction between the target site and miRNA53. Eleven miRNAs were found to be participated in cleavage of the target transcript, although six miRNAs supposedly inhibit the translation of target genes. These miRNAs were previously identified as novel or conserved miRNAs by Yu et al.54 and Jiang et al.55 in B. rapa L.and B. comparstis ssp. chinensis, respectively. Former research has shown that some of these miRNA families are greatly preserved in Brassicaceae or other plant species, located and expressed in leaves, pollen, roots or flower, with ancient functions in heat stress response (bra-miR5726, bra-miR5712 and bra-miR5716)54,56, regulation of target genes related to plant development (i.e., bra-miR156a/b/d-3p, bra-miR824, and bra-miR391-5p)55, somatic embryogenesis in Dimocarpus longan57, Brassica-specific hormone signal transduction pathway (i.e., bra-miR162-3p), drought stress tolerance in tomato (i.e., miR160a and miR9552b)58 and response to Turnip mosaic virus (i.e., bra-miR1885a and bra-miR5717)57. The function of the remaining novel and conserved miRNAs is not known yet, which requires further experimental elucidation.
In-silico functional relationship network of BrCNGC proteins
A theoretical protein–protein interaction was constructed with the STRING program to recognise the relations among unlike BoCNGC proteins59. The interaction network of first shell of interactors presented that thirteen BrCNGCs were part of various protein–protein interaction networks (Supplementary Fig. S12). Among these, seven proteins, namely BrCNGC2, 14–18 and interact with ubiquitin3 protein (Bra009542), detected by Affinity Capture-MS assay. It is reported that Polyubiquitin chain upon covalent binding to target protein governs proteolysis, DNA damage tolerance and other processes60. In another association, BrCNGC29 interact with Constitutive Photomorphogenic 1, experimentally determined by biochemical data from psi-mi (fluorescent resonance energy transfer) assay and two-hybrid assay during former research on Arabidopsis CNGCs. The functional annotation showed that COP1 serve as a negative regulator of photomorphogenesis in Arabidopsis61. Similarly, BrCNGC2 interacted with multiple proteins including BrCNGC18 and Bra00322 (a truncated CNGC gene), whose genes probably have correlated expression.
Functional analyses of BrCNGCs by transcriptome-based expression profiling
Expression patterns in different plant parts and wounding stress
Scrutinising the steady-state expression patterns of BrCNGC genes in six tissues (i.e., root, stem, flower, silique, leaf, and callus) was performed via Illumina RNA-sequencing data from the Gene Expression Omnibus (GEO) database database. Out of the 29 BrCNGCs, fifteen were expressed at moderately high levels (fragments per kilobase of exon model per million mapped reads value > 1) in at least one tissue, including ten in silique, eleven in calli, twelve in the roots and stem, and fourteen in leaves and flowers. The remaining genes either displayed lowest transcript accumulation or did not express in any tissue (Fig. 5; Supplementary Table S6). An additional investigation revealed that BrCNGC21 was the highest expressed genes, particularly in flowers and silique, suggesting they may be vital for Brassica species development. Amongst the other genes, BrCNGC4 was greatly expressed in leaves, BrCNGC7 in stem and roots, although BrCNGC16 was greatly expressed in calli. Greater expression in silique and calli suggest the expression of these genes is induced by wounding.

Heatmap showing the transcript abundance of BrCNGC genes in different development tissues of Brassica rapa L. The gene names and cluster tree are indicated on the left side of the figure. Normalized gene expression (FPKM) is expressed in log2 ratio, with yellow colors indicating lower accumulation of transcripts, and green colors indicating higher accumulation of the gene transcripts. The intensity of transcript abundance is indicated as white histograms within the heatmap.
Our data suggest that BrCNGC genes in different tissues expressed differently, and that several genes are induced by wounding22. Highly expressed genes in certain tissues indicated some functional preservation, while others showing functional dissimilarities62,63.
Expression patterns in response to hormonal stress
RNA-Seq technology allows a better understanding of the regulation of the important genes in the secondary metabolite biosynthetic pathways in plants, including Brassica64. Methyl jasmonate (MeJA) is one such plant hormone that is used in diverse developmental pathways and defense in plants65. We determined the expression profiles of 29 BrCNGCs in the leaves of B. rapa, exposed to 0.2 mM of MeJA (Supplementary Table S7). The calculated fold-change data showed that fourteen genes were up-regulated at 8–10th leaf stage, seven genes were down-regulated, while the remaining genes didn’t show low transcript abundance compared to control (Fig. 6a). Among these, BrCNGC13 showed maxim level of expression, which was up-regulated > 5.8-fold compared to unstressed control. On other hand, BrCNGC18 showed maximum negative response, which was—ninefold down-regulated compared to control. This pattern was followed by BrCNGC25 and BrCNGC29 respectively. These results indicated that the transcriptional responses of CNGCs along with other signal transduction pathway genes are regulated by MeJA66,67.

Dynamic expression profiles of BrCNGC genes in Brassica rapa L. plants, subjected to different stress types. (a) Exogenous hormone (0.2 mM of MeJA). (b) Bacterial pathogen (Psm) and elicitor flagellin (Flg22). (c) P. brassicae in clubroot resistant and susceptible Cabbage lines at 0, 12 72 and 96 h after inoculation. (d) Trace element stress represented by cadmium excess (CdE), iron deficiency (FeD), zinc excess (ZnE) and deficiency (ZnD), respectively. The final relative expression level of each transcript shown in this figure is calculated as fold change compared to controls/mocks, where threshold > 0 indicate up-regulation and threshold < 0 show down-regulation.
Expression patterns in response to bacterial pathogen and elicitor stress
Phytoalexins are antimicrobial substances produced by plants to elicit resistance against pathogen infection68. Most of the phyloalexin biosynthesis pathways are reported to be conserved across the B. rapa L. cultivars, Chiifu and Rapid Cycling (RCBr). Using illumina RNA-sequencing, Klein et al.69 observed that some of phyloalexin biosynthesis pathways are activated by infiltration with the Pseudomonas syringae pv. maculicola (Psm) and oligopeptide epitope of bacterial flagellin (flg22). Our search of the transcriptome data revealed the expression profiles of BrCNGC genes in the leaves of 15 days old RCBr plants, infiltrated by Psm and flg22. The FPKM values of 29 BrCNGC genes are shown in Supplementary Table S8. Most of the BrCNGC genes were expressed at higher levels after 9 h post-infiltration, including twenty-two genes in response to flg22, and twenty-one in response to Psm (Fig. 6b). Among these, > 18 BrCNGC genes were mutually expressed under both treatments, four expressed differentially, while seven genes didn’t show any expression compared to uninfected controls. Compared with their mock treatments, the expression of ten genes was increased and eleven decreased in response to Psm. The maximum responses were noted for BrCNGC27 (> tenfold up-regulation) and BrCNGC20 (– sixfold down-regulation), respectively. On other hand, the expression of thirteen genes was increased and nine decreased in response to flg22, with notable responses shown by BrCNGC12 (> sixfold up-regulation) and BrCNGC19 (i.e.,—7.2-fold down-regulation) respectively. The results showed that three duplicated gene pairs (i.e., BrCNGC-22/27, 25/28 and 26/29) has similar expression trend (Fig. 6b). These results indicate that various CNGCs may be involved plant defense against bacterial pathogens34.
Expression patterns in response to clubroot pathogen Plasmodiophora brassicae
Plasmodiophora brassicae is among the most common pathogens worldwide, which cause clubroot disease in Brassica crops70. In a latest study, Chen et al.71 profiled the transcriptomes of the roots from two near-isogenic lines (NILs) of B. rapa L., namely clubroot-resistant and clubroot-susceptible. This RNA-seq library (i.e., GSE74044) contained the expressions of 26 BrCNGCs in 30-days old B. rapa L. NILs inoculated with P. brassicae, and the data collected after 0, 12, 72 and 96 h after inoculation (Supplementary Table S9). The missing profiles of the remaining three genes (i.e., BrCNGC2, 8, and 14), might be due to no expression at all, or these genes had spatial and temporal expression patterns35. As shown in fig.ure6c, almost similar expression trends were observed between two NILs, where 17 to 19 genes were up-regulated, and five or six genes were down-regulated at one or other time point. Five genes, including BrCNGC19, 20, 22, 25 and 26, showed irregular expression between two cultivars at different time points. Comparatively, maximum level of expression was noted for BrCNGC9, which peaked in both NILs at all-time points (~ 37 to 44-folds), while, maximum negative responses was shown by BrCNGC25, which was—44-fold down-regulated in clubroot-susceptible at 96 hai. Among others, the transcripts of all genes, except BrCNGC-6, 15, 20, and 23–25, were up-regulated, showing that some of BrCNGCs can be further explored to understand their mechanism to facilitate resistance to P. brassicae.
Expression patterns in response to trace elements stress
Trace elements are essential for human nutrients to fulfill their metabolic requirements72. Among these trace elements, iron (Fe) and zinc (Zn) are mainly significant, because their deficiency cause serious health and nutritional problems in human population73. On the other hand, Cadmium (Cd) is a toxic element found in the soil, which cause severe toxicity in plants, animals and humans74. It is documented fact that the excess of zinc intake also cause toxicity, which can be more harmful to the plants, compared to Zn deficiency75. Taking advantage of recently published transcriptome data76, we investigated the expression patterns of BrCNGC genes in leaves of B. rapa L. plants cultivated under Cd excess (CdE), Fe deficiency (FeD), and Zn deficiency (ZnD) and excess (ZnE) conditions (Supplementary Table S10). Compared to control, seven genes were up-regulated under CdE, eight under FeD, twelve under ZnD, and eight genes were up-regulated under ZnE condition, respectively (Fig. 6d). On the contrary, nine genes were down-regulated under CdE, eight under FeD, six under ZnD, and eight genes were down-regulated under ZnE, respectively. Some of the multi-copy genes, such as BrCNGC26 and BrCNGC29, showed similar trend under ZnD stress, while other gene pairs exhibited differential patterns. These observations are agreement to the findings of Li et al.76. The data showed that some of BrCNGC family genes are definitely involved in trace elements response, and further experiments will clarify their individual roles and help in improving environmental adaptability in B. rapa L.
Methods
Genome-wide identification of CNGC proteins
The identify CNGC gene family in B. rapa L., the protein sequences of twenty Arabidopsis CNGCs were collected from TAIR1077 and BLAST searched against target proteomes in BRAD database78, using built-in BLASTP search. The matching protein sequences of target species were retrieved and analyzed in SMART79, Pfam80 and Motif search service on GenomeNet for domain analysis. Finally, the target protein sequences comprising cNMP-binding (IPR000595) and ion transport (PF00520) domains were recognized as candidate CNGCs and manually checked for the presence plant CNGC-specific consensus motif within the cNMP-binding region14. The newly identified CNGC genes were named according to standard nomenclature (i.e., taxonomic initials such Br for B. rapa L.) and phylogenetic positions.
Multiple sequence alignment and phylogenetic analysis
ClustalX 2.0 program was performed for Multiple sequence alignments of the BrCNGC proteins81 and were observed by GeneDoc82. MEGA software version 6.0, was used for phylogenetic tree construction83. For identification purposes, the BrCNGC proteins were individually aligned with AtCNGCs and phylogeny performed. Multiple sequence alignments based on the CNGC proteins from both species were used for combined rooted tree by using Amborella trichopoda CNGC (AMTR_s00210p00019190) as outgroup.
Characterization and properties of BrCNGCs
The data about gene and protein lengths, their chromosome locations and positional information of the CNGCs were obtained from BRAD database. The ProtParam tool was used to study the amino acid properties BrCNGC proteins84. The ScanProsite tool was used search the post-translational modifications sites48.
Chromosomal mapping, gene duplication and syntenic analysis
The positional information from BRAD database was used for genomic mapping of CNGC genes on B. rapa L. chromosomes by using R script. The tandem and segmental duplications were analyzed by PGDD85 and PTGBase39. The synteny relationship between BrCNGCs, AtCNGCs and BoCNGCs were assessed in Bolbase86, and mapped in a circos plot using R studio87.
Conserved motif composition and Gene structure
To predict the gene structures, we used Gene Structure Display Server (GSDS 2.0)88. To find conserved motifs in the CNGC protein sequences, we used the MEME and MAST motif discovery tools with default parameters44. The annotation of the motifs were performed in Pfam program45.
The miRNA target sites and protein–protein interaction
The microRNA sequences of B. rapa L. were collected from miRBase database89 and submitted to psRNATarget server52 for miRNA’s target sites prediction within the BrCNGC genes. Each of these miRNAs were searched online to find their experimental proof, function and related literature. The protein–protein interaction of BrCNGC proteins was constructed in STRING v1059, by using the CNGC protein sequences as reference.
Data sources and expression of BrCNGC genes
For expression profiling of BrCNGC genes in different plant tissues, the RNA-seq data placed in GEO database (GSE43245) was used38,86. For gene expression against different stress treatments, the expression data (GSE69785) of 15 days old RCBr plants infiltrated with Psm and flg2269, GSE74044 for expression in the roots of 30-day old NILs at 0, 12 , 72 and 96 h after inoculation of P. brassicae71, GSE51363 for expression in the leaves of B. rapa L. subsp. pekinensis, exposed to 0.2 mM MeJA at 8–10 leaf stage, and GSE55264 for expression in the leaves of 14 days plants exposed to Fe deficiency (0.05 µM; Normal = 3 µM), Zn deficiency (0.005 µM; Normal = 2 µM), Zn excess (50 µM; Normal = 2 µM) and Cd excess (1 µM; Normal = without Cd) for 7 days was used. Transcript abundance was calculated as FPKM and the values were log2 transformed. Data were plotted in heat maps generated in R studio90. For abiotic and biotic stress, we used a fold-change method, where the threshold of ≥ 0 defines a gene as “positively expressed/up-regulated” and threshold ≤ 0 as “negatively expressed/down-regulated”, compared to FPKM values in control treatments.
Conclusion
This work is the first wide-ranging and systematic study of CNGC gene family in B. rapa L. This work identifies and fills the remaining gaps in literature, and present a clearer picture about plant CNGCs in general, and crucifers in particular. Here, we have tried to explore each and every aspect of BrCNGC gene family, from genes to protein, including gene structure, motif composition, miRNA target sites, post-translational modification sites, protein interaction network, GO-term prediction and orthologous relationship etc. The phylogenetic and synteny analyses will help in understanding the evolutionary patterns, and diversification and/or expansion of CNGC family genes in complex ancient polyploids (e.g., B. rapa/B. oleracea), whose genomes have undergone multiple duplication and reshuffling events. Additionally, this work will contribute to further clarify the functions of differentially expressed candidate BrCNGC genes through cloning, and to investigate their roles in the regulation of cascade pathways, plant development and stress tolerance in B. rapa L.
Data availability
The raw sequence datasets generated and analyzed during the current study are available through BRAD database (http://brassicadb.org/brad/). The gene expression data analyzed during the current study are available at GEO database with accession numbers: GSE43245, GSE69785, GSE74044, GSE51363 and GSE55264.
Abbreviations
- B. rapa L.:
-
Brassica rapa L
- CNGC:
-
Cyclic nucleotide-gated ion channel
- cNMPs:
-
Cyclic nucleotide monophosphates
- flg22:
-
Flagellin
- Psm :
-
Pseudomonas syringae Pv. Maculicola
- NILs:
-
Near-isogenic lines
- CaM:
-
Calmodulin
References
Mäser, P. et al. Phylogenetic relationships within cation transporter families of Arabidopsis. Plant Physiol. 126, 1646–1667 (2001).
Borsics, T., Webb, D., Andeme-Ondzighi, C., Staehelin, L. A. & Christopher, D. A. The cyclic nucleotide-gated calmodulin-binding channel AtCNGC10 localizes to the plasma membrane and influences numerous growth responses and starch accumulation in Arabidopsis thaliana. Planta 225, 563–573 (2007).
Christopher, D. A. et al. The cyclic nucleotide gated cation channel AtCNGC10 traffics from the ER via Golgi vesicles to the plasma membrane of Arabidopsis root and leaf cells. BMC Plant Biol. 7, 48 (2007).
Yuen, C. C. & Christopher, D. A. The group IV-A cyclic nucleotide-gated channels, CNGC19 and CNGC20, localize to the vacuole membrane in Arabidopsis thaliana. AoB Plants 5, plt012 (2013).
Charpentier, M. et al. Nuclear-localized cyclic nucleotide–gated channels mediate symbiotic calcium oscillations. Science 352, 1102–1105 (2016).
Jha, S. K., Sharma, M. & Pandey, G. K. Role of cyclic nucleotide gated channels in stress management in plants. Curr. Genom. 17, 315–329. https://doi.org/10.2174/1389202917666160331202125 (2016).
Ma, W. & Berkowitz, G. A. Cyclic nucleotide gated channel and Ca2+-mediated signal transduction during plant senescence signaling. Plant Signal. Behav. 6, 413–415 (2011).
Ma, W. et al. Leaf senescence signaling: The Ca2+-conducting Arabidopsis cyclic nucleotide gated channel2 acts through nitric oxide to repress senescence programming. Plant Physiol. 154, 733–743 (2010).
Zelman, A. K., Dawe, A., Berkowitz, G. A. & Gehring, C. Evolutionary and structural perspectives of plant cyclic nucleotide-gated cation channels. Front. Plant Sci. 3, 95 (2012).
Guo, K. M., Babourina, O., Christopher, D. A., Borsics, T. & Rengel, Z. The cyclic nucleotide-gated channel, AtCNGC10, influences salt tolerance in Arabidopsis. Physiol. Plant. 134, 499–507 (2008).
Almoneafy, A. A. et al. Tomato plant growth promotion and antibacterial related-mechanisms of four rhizobacterial Bacillus strains against Ralstonia solanacearum. Symbiosis 63, 59–70 (2014).
Chen, J. et al. Genomic characterization, phylogenetic comparison and differential expression of the cyclic nucleotide-gated channels gene family in pear (Pyrus bretchneideri Rehd). Genomics 105, 39–52. https://doi.org/10.1016/j.ygeno.2014.11.006 (2015).
Saand, M. A., Xu, Y.-P., Li, W., Wang, J.-P. & Cai, X.-Z. Cyclic nucleotide gated channel gene family in tomato: genome-wide identification and functional analyses in disease resistance. Front. Plant Sci. 6, 303 (2015).
Zelman, A. K., Dawe, A. & Berkowitz, G. A. in Cyclic Nucleotide Signaling in Plants: Methods and Protocols (ed Chris Gehring) 207–224 (Humana Press, 2013).
Nawaz, Z. et al. Genome-wide identification, evolution and expression analysis of cyclic nucleotide-gated channels in tobacco (Nicotiana tabacum L). Genomics 111, 142–158. https://doi.org/10.1016/j.ygeno.2018.01.010 (2019).
Kakar, K. U. et al. Comprehensive genomic analysis of the CNGC gene family in Brassica oleracea: novel insights into synteny, structures, and transcript profiles. BMC Genomics 18, 869 (2017).
DeFalco, T. A. et al. Multiple calmodulin-binding sites positively and negatively regulate arabidopsis cyclic nucleotide-gated channel12. Plant Cell 1, 1 (2016).
Newton, R. P. & Smith, C. J. Cyclic nucleotides. Phytochemistry 65, 2423–2437. https://doi.org/10.1016/j.phytochem.2004.07.026 (2004).
Kaplan, B., Sherman, T. & Fromm, H. Cyclic nucleotide-gated channels in plants. FEBS Lett. 581, 2237–2246. https://doi.org/10.1016/j.febslet.2007.02.017 (2007).
Chin, K., DeFalco, T. A., Moeder, W. & Yoshioka, K. The Arabidopsis cyclic nucleotide-gated ion channels AtCNGC2 and AtCNGC4 work in the same signaling pathway to regulate pathogen defense and floral transition. Plant Physiol. 163, 611–624 (2013).
Finka, A., Cuendet, A. F. H., Maathuis, F. J., Saidi, Y. & Goloubinoff, P. Plasma membrane cyclic nucleotide gated calcium channels control land plant thermal sensing and acquired thermotolerance. Plant Cell 24, 3333–3348 (2012).
Aab, K. U. K., Nawaz, Z. & Ahmed, J. Recent trend of genome-wide multigene family analysis and their role in plant drought tolerance. Ann. Agric. Crop Sci. 4, 1046 (2019).
Warwick, S. I., Francis, A. & Al-Shehbaz, I. A. Brassicaceae: species checklist and database on CD-Rom. Plant Syst. Evol. 259, 249–258 (2006).
Wang, X. et al. The genome of the mesopolyploid crop species Brassica rapa. Nat. Genet. 43, 1035–1039 (2011).
Lim, T. in Edible Medicinal and Non Medicinal Plants 777–788 (Springer, 2015).
Haytowitz, D. B., Pehrsson, P. R. & Holden, J. M. The national food and nutrient analysis program: a decade of progress. J. Food Compost. Anal. 21, S94–S102 (2008).
Kris-Etherton, P. M. et al. Bioactive compounds in foods: their role in the prevention of cardiovascular disease and cancer. Am. J. Med. 113, 71–88 (2002).
Kristal, A. R. & Lampe, J. W. Brassica vegetables and prostate cancer risk: a review of the epidemiological evidence. Nutr. Cancer 42, 1–9 (2002).
Wang, L. I. et al. Dietary intake of Cruciferous vegetables, Glutathione S-transferase (GST) polymorphisms and lung cancer risk in a Caucasian population. Cancer Causes Control 15, 977–985 (2004).
Gauss, R., Seifert, R. & Kaupp, U. B. Molecular identification of a hyperpolarization-activated channel in sea urchin sperm. Nature 393, 583–587 (1998).
Henn, D. K., Baumann, A. & Kaupp, U. B. Probing the transmembrane topology of cyclic nucleotide-gated ion channels with a gene fusion approach. Proc. Natl. Acad. Sci. 92, 7425–7429 (1995).
Saand, M. A. et al. Phylogeny and evolution of plant cyclic nucleotide-gated ion channel (CNGC) gene family and functional analyses of tomato CNGCs. DNA Res. 22, 471–483 (2015).
Li, Q., Yang, S., Ren, J., Ye, X. & Liu, Z. Genome-wide identification and functional analysis of the cyclic nucleotide-gated channel gene family in Chinese cabbage. Biotech 9, 1–114 (2019).
Nawaz, Z., Kakar, K. U., Saand, M. A. & Shu, Q.-Y. Cyclic nucleotide-gated ion channel gene family in rice, identification, characterization and experimental analysis of expression response to plant hormones, biotic and abiotic stresses. BMC Genom. 15, 1 (2014).
Song, X.-M. et al. Genome-wide analysis of the GRAS gene family in Chinese cabbage (Brassica rapa ssp. pekinensis). Genomics 103, 135–146 (2014).
Nagaharu, U. Genome analysis in Brassica with special reference to the experimental formation of B. napus and peculiar mode of fertilization. Jpn J Bot 7, 389–452 (1935).
Parkin, I. A. P. et al. Transcriptome and methylome profiling reveals relics of genome dominance in the mesopolyploid Brassica oleracea. Genome Biol. 15, R77–R77. https://doi.org/10.1186/gb-2014-15-6-r77 (2014).
Liu, S. et al. The Brassica oleracea genome reveals the asymmetrical evolution of polyploid genomes. Nat. Commun. 5, 1. https://doi.org/10.1038/ncomms4930 (2014).
Liang, Y. et al. Genome-wide identification, structural analysis and new insights into late embryogenesis abundant (LEA) gene family formation pattern in Brassica napus. Sci. Rep. 6, 1 (2016).
Duan, W. et al. Patterns of evolutionary conservation of ascorbic acid-related genes following whole-genome triplication in Brassica rapa. Genome Biol. Evol. 7, 299–313 (2015).
Cheng, F. et al. Deciphering the diploid ancestral genome of the mesohexaploid Brassica rapa. Plant Cell 25, 1541–1554 (2013).
Xu, G., Guo, C., Shan, H. & Kong, H. Divergence of duplicate genes in exon–intron structure. Proc. Natl. Acad. Sci. 109, 1187–1192 (2012).
Wang, L. et al. Genome-wide identification of WRKY family genes and their response to cold stress in Vitis vinifera. BMC Plant Biol. 14, 1 (2014).
Bailey, T. L. et al. MEME SUITE: Tools for motif discovery and searching. Nucl. Acids Res. 37, W202–W208 (2009).
Finn, R. D. et al. Pfam: the protein families database. Nucl. Acids Res. 42, D222–D230 (2013).
Duan, G. & Walther, D. The roles of post-translational modifications in the context of protein interaction networks. PLoS Comput. Biol. 11, e1004049 (2015).
Webster, D. E. & Thomas, M. C. Post-translational modification of plant-made foreign proteins; glycosylation and beyond. Biotechnol. Adv. 30, 410–418 (2012).
De Castro, E. et al. ScanProsite: detection of PROSITE signature matches and ProRule-associated functional and structural residues in proteins. Nucl. Acids Res. 34, W362–W365 (2006).
Rogers, S., Wells, R. & Rechsteiner, M. Amino acid sequences common to rapidly degraded proteins: the PEST hypothesis. Science 234, 364–368 (1986).
Liu, B., Li, J. & Cairns, M. J. Identifying miRNAs, targets and functions. Brief. Bioinform. 15, 1–19. https://doi.org/10.1093/bib/bbs075 (2014).
Witkos, T. M., Koscianska, E. & Krzyzosiak, W. J. Practical aspects of microRNA target prediction. Curr. Mol. Med. 11, 93–109. https://doi.org/10.2174/156652411794859250 (2011).
Dai, X. & Zhao, P. X. psRNATarget: A plant small RNA target analysis server. Nucleic Acids Res. 39, W155–W159 (2011).
Marín, R. M. & Vaníček, J. Efficient use of accessibility in microRNA target prediction. Nucl. Acids Res. 39, 19–29 (2011).
Yu, X. et al. Identification of conserved and novel microRNAs that are responsive to heat stress in Brassica rapa. J. Exp. Bot. 63, 1025–1038. https://doi.org/10.1093/jxb/err337 (2011).
Jiang, J., Lv, M., Liang, Y., Ma, Z. & Cao, J. Identification of novel and conserved miRNAs involved in pollen development in Brassica campestris ssp. chinensis by high-throughput sequencing and degradome analysis. BMC Genom. 15, 146–146. https://doi.org/10.1186/1471-2164-15-146 (2014).
Yu, X. et al. Global analysis of cis-natural antisense transcripts and their heat-responsive nat-siRNAs in Brassica rapa. BMC Plant Biol. 13, 208–208. https://doi.org/10.1186/1471-2229-13-208 (2013).
Wang, Z. et al. Genome-wide identification of turnip mosaic virus-responsive microRNAs in non-heading Chinese cabbage by high-throughput sequencing. Gene 571, 178–187 (2015).
Candar-Cakir, B., Arican, E. & Zhang, B. Small RNA and degradome deep sequencing reveals drought-and tissue-specific micrornas and their important roles in drought-sensitive and drought-tolerant tomato genotypes. Plant Biotechnol. J. 14, 1727–1746. https://doi.org/10.1111/pbi.12533 (2016).
Szklarczyk, D. et al. STRING v10: protein–protein interaction networks, integrated over the tree of life. Nucl. Acids Res. 43, D447–D452 (2014).
Li, W. & Ye, Y. Polyubiquitin chains: functions, structures, and mechanisms. Cell. Mol. Life Sci.: CMLS 65, 2397–2406. https://doi.org/10.1007/s00018-008-8090-6 (2008).
Bauer, D. et al. Constitutive photomorphogenesis 1 and multiple photoreceptors control degradation of phytochrome interacting factor 3, a transcription factor required for light signaling in arabidopsis. Plant Cell 16, 1433–1445. https://doi.org/10.1105/tpc.021568 (2004).
Ali, E. et al. Bioinformatics study of Tocopherol biosynthesis pathway genes in Brassica rapa. Int. J. Curr. Microbiol. App. Sci 4, 721–732 (2015).
Kakar, K. U. et al. Evolutionary and expression analysis of CAMTA gene family in Nicotiana tabacum yielded insights into their origin, expansion and stress responses. Sci. Rep. 8, 10322. https://doi.org/10.1038/s41598-018-28148-9 (2018).
Miao, L. et al. Transcriptome analysis of stem and globally comparison with other tissues in Brassica napus. Front. Plant Sci. 7, 1 (2016).
Wang, R., Xu, S., Wang, N., Xia, B. & Jiang, Y. Transcriptome analysis of secondary metabolism pathway, transcription factors and transporters in response to methyl jasmonate in Lycoris aurea. Front. Plant Sci. 7, 1971 (2016).
Yi, G.-E. et al. Exogenous Methyl Jasmonate and Salicylic Acid Induce Subspecies-Specific Patterns of Glucosinolate Accumulation and Gene Expression in Brassica oleracea L. Molecules 21, 1417 (2016).
Zhai, X. et al. The regulatory mechanism of fungal elicitor-induced secondary metabolite biosynthesis in medical plants. Crit. Rev. Microbiol. 1, 1–24 (2016).
To, Q. Unveiling the first indole-fused thiazepine: structure, synthesis and biosynthesis of cyclonasturlexin, a remarkable cruciferous phytoalexin. Chem. Commun. 52, 5880–5883 (2016).
Klein, A. P. & Sattely, E. S. Two cytochromes P450 catalyze S-heterocyclizations in cabbage phytoalexin biosynthesis. Nat. Chem. Biol. 11, 837–839. https://doi.org/10.1038/nchembio.1914 (2015).
Rolfe, S. A. et al. The compact genome of the plant pathogen Plasmodiophora brassicae is adapted to intracellular interactions with host Brassica spp. BMC Genom. 17, 1 (2016).
Chen, J., Pang, W., Chen, B., Zhang, C. & Piao, Z. Transcriptome analysis of Brassica rapa near-isogenic lines carrying clubroot-resistant and –susceptible alleles in response to plasmodiophora brassicae during early infection. Front. Plant Sci. 6, 1183. https://doi.org/10.3389/fpls.2015.01183 (2015).
Welch, R. M. & Graham, R. D. Breeding for micronutrients in staple food crops from a human nutrition perspective. J. Exp. Bot. 55, 353–364 (2004).
Petry, N., Olofin, I., Boy, E., Donahue Angel, M. & Rohner, F. The effect of low dose iron and zinc intake on child micronutrient status and development during the first 1000 days of life: A systematic review and meta-analysis. Nutrients 8, 773 (2016).
Clemens, S., Aarts, M. G., Thomine, S. & Verbruggen, N. Plant science: the key to preventing slow cadmium poisoning. Trends Plant Sci. 18, 92–99 (2013).
Wu, J. et al. Characterization of natural variation for zinc, iron and manganese accumulation and zinc exposure response in Brassica rapa L. Plant Soil 291, 167–180 (2007).
Thakur, N. et al. Enhanced whitefly resistance in transgenic tobacco plants expressing double stranded RNA of v-ATPase A gene. PLoS One 9, e87235 (2014).
Lamesch, P. et al. The Arabidopsis Information Resource (TAIR): improved gene annotation and new tools. Nucl. Acids Res. 40, D1202–D1210 (2012).
Cheng, F. et al. BRAD, the genetics and genomics database for Brassica plants. BMC Plant Biol. 11, 1 (2011).
Letunic, I., Doerks, T. & Bork, P. SMART: recent updates, new developments and status in 2015. Nucl. Acids Res. 43, D257–D260 (2015).
Finn, R. D. et al. The Pfam protein families database: towards a more sustainable future. Nucl. Acids Res. 44, D279–D285 (2016).
Larkin, M. A. et al. Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948 (2007).
Nicholas, K. B. GeneDoc: Analysis and visualization of genetic variation. Embnew. news 4, 14 (1997).
Tamura, K., Stecher, G., Peterson, D., Filipski, A. & Kumar, S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729 (2013).
Gasteiger, E. et al. in The Proteomics Protocols Handbook (ed John M. Walker) 571–607 (Humana Press, 2005).
Lee, T.-H., Tang, H., Wang, X. & Paterson, A. H. PGDD: A database of gene and genome duplication in plants. Nucl. Acids Res. 41, D1152–D1158 (2013).
Tong, C. et al. Comprehensive analysis of RNA-seq data reveals the complexity of the transcriptome in Brassica rapa. BMC Genom. 14, 1 (2013).
Allaire, J. RStudio: integrated development environment for R. Boston, MA 770, 165–171 (2012).
an upgraded gene feature visualization server. Hu, B. et al. GSDS 2.0. Bioinformatics 31, 1296–1297 (2014).
Kozomara, A. & Griffiths-Jones, S. miRBase: annotating high confidence microRNAs using deep sequencing data. Nucl. Acids Res. 42, D68–D73 (2014).
Studio, R. RStudio: integrated development environment for R (RStudio Inc, 2012).
Acknowledgements
We are grateful to Zhejiang University for providing us support.
Author information
Authors and Affiliations
Contributions
K.U.K. and A.A.B. designed and conceptualized this study. A.A.B. collected the sequence data and performed bioinformatics work with A.M.R. and S.S.A.R. S.U. and S.K. performed sequence alignments, synteny, phylogenetic analysis and prepared figures. Z.N. and H.Z. contributed in data visualization, drafting, formatting and language editing of the manuscript along with G.K.M. A.M.R and G.K.M. analyzed gene expression data and helped in revision with others. All authors commented at each stage. K.U.K supervised the study.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Baloch, A.A., Raza, A.M., Rana, S.S.A. et al. BrCNGC gene family in field mustard: genome-wide identification, characterization, comparative synteny, evolution and expression profiling. Sci Rep 11, 24203 (2021). https://doi.org/10.1038/s41598-021-03712-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-03712-y
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
