


 Figure 1: An illustration of how epigenetic mechanisms can affect health
Figure 1: An illustration of how epigenetic mechanisms can affect health















« Prev Next »
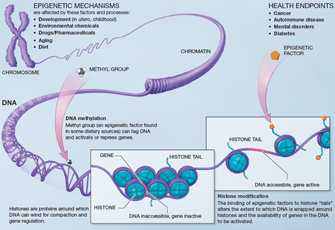
National Institutes of Health.
Having one version of a gene rather than another may render a person more likely to develop cancer or diabetes, or perhaps to be overweight rather than slender. However, identical twins, who inherit exactly the same set of genes from their parents, can end up with different physical characteristics, behaviors, and even predispositions to disease. How is this possible? In short, such differences arise because various epigenetic factors influence whether certain genes are active or not, as well as when and where these genes become active.
DNA methylation and histone modification (Figure 1) are two primary examples of processes that result in epigenetic modifications to the genome. Changes linked to both of these processes accumulate throughout a person's life and can be passed on from one generation to the next. Epigenetic mechanisms regulate many important biological processes, such as those responsible for cell division and cell differentiation. These mechanisms are also involved in many diseases, including cancer and diabetes.
Although a person's complement of genes—in other words, his or her genome—remains essentially the same from birth onward (except for the occurrence of mutations that can change the function of genes), different environmental exposures during development and throughout a person's life chemically modify DNA and the proteins bound to it. Thus, even identical genomes from twins can be subject to distinct epigenetic changes. For instance, when one group of researchers analyzed the chromosomes of identical twins, they found differences in the extent to which the DNA was tagged by methyl groups—a process known as DNA methylation, which plays a role in regulating the transcription of genes. These researchers also found that each individual's histones, or the proteins around which DNA winds when it is compacted into chromosomes, carried different chemical tags. These tags are thought to alter the extent to which DNA is wrapped around the histones, thereby affecting the availability of genes for activation. The researchers reasoned that these changes accounted for, at least in part, the various physical and behavioral differences between identical twins (Fraga et al., 2005).
Thus, the field of epigenetics focuses on the study of heritable changes rather than mutations in the primary DNA sequence. The term "epigenetics" was coined in 1940 by biologist Conrad Waddington, who defined it as "the interactions of genes with their environment which bring the phenotype into being" (Waddington, 1940). Over time, the field of epigenetics gave rise to that of epigenomics, which is the study of epigenetic modifications across an individual's entire genome. Epigenomics has only become possible in recent years because of the advent of various sequencing tools and technologies, such as DNA microarrays, cheap whole-genome resequencing, and databases for studying entire genomes (Bonetta, 2008). Why Is Epigenomics Important?
The human genome contains approximately 25,000 genes that encode all the proteins and other molecules that make up an individual. The epigenome is important to this vast collection of nucleotides in that it provides instructions for when some of these proteins are produced, as well as in which cells or tissues production takes place. In the words of researcher Dana Dolinoy (2007), "A useful analogy is to think of the epigenome as the software that directs the genomic hardware of a computer." Another way to think of the epigenome is to picture it as DNA "formatting"—here, if our genome is referred to as the book of life, written in A's, T's, G's, and C's, then the epigenome represents the spaces and punctuation that format the text. Epigenetic marks are literally made on the DNA itself, or on the proteins that package it in the nucleus. Epigenetics and epigenomics help explain the mechanism by which our environment affects our phenotype.
Similar to the recently completed Human Genome Project, which decoded the complete sequence of the human genome, several large-scale projects and consortia are now underway in an attempt to characterize the many human epigenomes (Jones et al., 2008). However, such projects will undoubtedly be more challenging than the Human Genome Project, because the epigenome differs over an individual's lifetime and is affected by several factors, including environmental chemicals, drugs and pharmaceuticals, aging, and diet. In addition, different tissues have different epigenetic marks across their genomes. Thus, epigenomics requires the identification of changes not only among different individuals, but also among different tissues, developmental stages, and disease types.
Studying DNA Methylation
One of the most studied epigenetic mechanisms is DNA methylation, particularly the process by which a methyl group is added to a cytosine residue, thereby transforming the cytosine into 5-methylcytosine. Currently, researchers have many methods for detecting such changes in methylation across the entire genome. Some of these approaches are described in the following sections.
Methylated DNA Immunoprecipitation
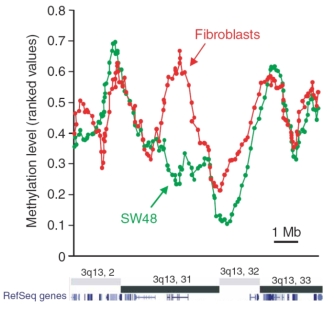
© 2005 Nature Publishing Group Weber, W. et al. Chromosome-wide and promoter-specific analyses identify sites of differential DNA methylation in normal and transformed human cells. Nature Genetics 37, 856 (2005). All rights reserved. 
One method employed in the study of methylation involves the use of antibodies to 5-methylcytosine; here, after cutting up genomic DNA, scientists can use these antibodies to pull out DNA fragments that contain methylated cytosine. The DNA that is bound to the antibodies is then purified, amplified, labeled with a fluorescent tag, and applied to the surface of a DNA microarray containing a set of probes from across the genome. When a fluorescently labeled piece of DNA binds to a particular probe in the microarray, it produces a signal. Scientists can then use the signal to determine where the probe maps within the genome, thereby revealing where DNA methylation has occurred.
Weber et al. (2005) were among the first investigators to use this technique—called methylated DNA immunoprecipitation or MeDIP—to find differences in methylation between normal and cancerous human cell lines. They determined that the methylation patterns in the two types of cells are similar, but they also noted defined regions on different chromosomes of differential methylation. Figure 2 shows the methylation of normal skin cells (specifically, fibroblasts, which are depicted in red) and of SW48 cancer cells (depicted in green). As shown in the figure, one region on the long arm of chromosome 3 (3q) has less methylation in the cancer cells. By looking at a genetic database (RefSeq) that tells where genes are located in the genome, the scientists determined that the region on chromosome 3q that is hypomethylated in the cancer cell line has few genes.
Other studies have shown that the promoter regions of genes become more methylated in cancer cells. In humans, cytosine often becomes methylated when it is next to a guanosine—this arrangement is known as a CpG dinucleotide. There are many CpG dinucleotides in the genome, and most of them are methylated. However, groups of unmethylated CpG dinucleotides often occur in the regulatory regions of genes and are referred to as CpG islands. Unmethylated CpG islands allow genes to be transcribed when the necessary transcription factors are present.
Comparative Methylation Hybridization, Bisulfite Treatment, and DNA Sequencing
Another approach for detecting differences in methylation that also uses microarrays is comparative methylation hybridization. Using this method, genomic DNA is digested with enzymes that are either sensitive or insensitive to methylation—in other words, they either will or will not cut a particular site if the DNA is methylated—before the DNA is applied to a microarray.
A third way to find methylated sites across the genome uses bisulfite treatment of DNA, which converts unmethylated cytosine into uracil. Bisulfite-treated DNA can be amplified using a version of polymerase chain reaction (PCR) that amplifies sequences containing 5-methylcytosines and not uracils—in other words, it amplifies those sequences that were not converted by the bisulfite (Clark et al., 1994). Bisulfite-treated DNA can also be hybridized to microarrays containing two sets of oligonucleotides, one of which is complementary to the unaltered methylated sequence, and the other which is complementary to the converted unmethylated sequence. Hybridization to one set of probes but not the other reveals where methylation occurs across the genome.
Of course, one problem associated with the use of microarrays is that they do not contain every single piece of DNA within the genome. Thus, to analyze the entire genome, scientists have started using a different approach: DNA sequencing. For instance, a collaboration between the Wellcome Trust Sanger Institute in the United Kingdom, the Center National de Genotypage in France, and the German company Epigenomics used this method in their analysis of methylation patterns on several regions of human chromosomes 6, 20, and 22 in 12 different tissues. The investigators found that regions that have been conserved throughout evolution show the greatest differences in DNA methylation. During their research, these scientists analyzed the sequence of 873 genes and discovered that these genes were differentially methylated in their regulatory regions (Eckhardt et al., 2006). More recently, Cokus et al. (2008) combined bisulfite treatment with new sequencing technologies to look at patterns of DNA methylation across the entire genome of the plant Arabidopsis thaliana.
Finding Patterns of Histone Modification
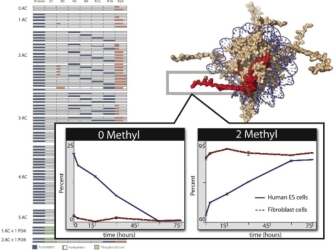
© 2008 Modified with permission from Joshua Coons. All rights reserved.
More recently, researchers have also started using the technique of mass spectrometry to determine modification patterns on different histones. For example, Joshua Coon's group at the University of Wisconsin-Madison used mass spectrometry to map all the modifications that occur in the 23 residues of the N-terminal tail of the histone H4 in human embryonic stem cells (Phanstiel et al., 2008). Figure 3 shows a map of all 74 histone H4 combination codes detected in human embryonic stem (ES) cells. Each box represents a potential site of modification, and coloration indicates the presence or absence of modifications. These mass spectrometry experiments show that the amino acid lysine at position 20 (K20) is the target of two methylation events.
The front panels in the figure show the effects on methylation once cells were treated with a chemical that induces differentiation. Specifically, levels of unmethylated histone H4 isoforms decreased in human ES cells during differentiation, while dimethylated isoforms increased. Note that the control human ES cells were characterized by a large percentage of unmethylated histone H4 (approximately 20%) and that upon 75 hours of treatment, this population was virtually eliminated in favor of dimethylation. The red line in the panel shows the results of this experiment using somatic cells (fibroblasts) exposed to the same differentiation agent. Note that no significant change was observed in this cell population.
The Epigenome and Disease
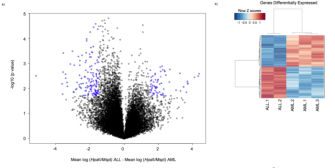
Creative Commons Figueroa, M. E., et al. An Integrative Genomic and Epigenomic Approach for the Study of Transcriptional Regulation. PLOS ONE 3 (3) 1882 (2008) doi:10.1371/journal.pone.0001882.
Today, many companies provide microarrays that contain gene promoters, CpG islands, or whole genomes so that scientists can look for methylation at these sites using MeDIP (Bonetta, 2008). Techniques like this allow scientists to study epigenetic changes across entire genomes, determining where these changes occur and under what circumstances. By combining such studies with those that determine the binding sites of transcription factors and other regulatory proteins across the genome, scientists will gain a better understanding of how epigenetic modifications occur and what their consequences are. Moreover, by determining patterns of methylation tags across the whole genome and of histone modifications in different types of cells and tissues, scientists will be able to find various "signatures" associated with particular biological processes or diseases states.
One example of such a signal exists in acute myeloid leukemia (AML) and acute lymphocytic leukemia (ALL). Scientists have compared the global methylation patterns from these two leukemias and found that they could easily be distinguished (Figure 4).
Epigenomic studies might also play a role in the development of new treatments for disease. Although epigenomic changes are heritable, drug treatments can potentially reverse them, opening the door for possible treatments. For instance, in one study using mice that had different coat colors depending on the amount of methylation in the DNA upstream of a particular gene, researchers found that giving pregnant mothers bisphenol A (BPA), a chemical widely used in the manufacture of plastic, increased DNA methylation in their offspring. This increased methylation was associated with higher body weight in the offspring. The scientists were able to change the effects of BPA on methylation and body weight in the offspring by feeding pregnant mice foods that contained a lot of methyl groups, such as foods rich in folic acid and soy protein (Dolinoy et al., 2007).
The Future of Epigenomic Research
Currently, several research groups are exploiting whole-genome approaches in an attempt to characterize the epigenome. Indeed, multiple different consortia aimed at the study of human epigenomics have formed in the past decade. These groups are working to identify, catalogue, and interpret genome-wide DNA methylation patterns of all human genes in all major tissues. Such collaborations are multinational and multidisciplinary, and they are a hallmark of twenty-first-century "-omics" research. Epigenomics is a new field with the potential to dissect nongenetic components of complex diseases. The hope is that with full genome sequence and epigenomic maps of the DNA methylation and modified histone landscapes, researchers will be able to tell exactly which genes are "turned on" and in which tissues. Integration of this massive amount of data promises to revolutionize our understanding of gene-environment interactions and offer new ways to diagnose and treat complex diseases.
References and Recommended Reading
Bonetta, L. Epigenomics: Detailed analysis. Nature 454, 795–798 (2008) doi:10.1038/454795a (link to article)
Callihan, P. A., & Feinberg, A. P. The emerging science of epigenomics. Human Molecular Genetics 15, R95–R101 (2006)
Clark, S. J., et al. High sensitivity mapping of methylated cytosines. Nucleic Acids Research 22, 2990–2997 (1994)
Cokus, S. J., et al. Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature 452, 215–219 (2008) (link to article)
Dennis, C. Epigenetics and disease: Altered states. Nature 421, 686–688 (2007) doi:10.1038/421686a (link to article)
Dolinoy, D. C. A Tale of Two Mice (video clip). NOVA website, http://www.pbs.org/wgbh/nova/sciencenow/3411/02.html (2007) (link to article)
Dolinoy, D. C., et al. Maternal nutrient supplementation counteracts bisphenol A-induced DNA hypomethylation in early development. Proceedings of the National Academy of Sciences 104, 13056–13061 (2007)
Eckhardt, F., et al. DNA methylation profiling of human chromosomes 6, 20, and 22. Nature Genetics 38, 1378–1385 (2006) doi:10.1038/ng1909 (link to article)
Feinberg, A. P., & Tycko, B. The history of cancer epigenetics. Nature Reviews Cancer 4, 143–153 (2004) doi:10.1038/nrc1279
Fraga, M. F., et al. Epigenetic differences arise during the lifetime of monozygotic twins. Proceedings of the National Academy of Sciences 102, 10604–10609 (2005)Jirtle, R. L., & Skinner, M. K. Environmental epigenomics and disease susceptibility. Nature Reviews Genetics 8, 253–262 (2007) doi:10.1038/nrg2045 (link to article)
Jones, P. A., & Baylin, S. B. The fundamental role of epigenetic events in cancer. Nature Reviews Genetics 3, 415–428 (2002) doi:10.1038/nrg816 (link to article)
Jones P. A., et al. Moving AHEAD with an international human epigenome project. Nature 454, 711–715 (2008) doi:10.1038/454711a (link to article)
Phanstiel, D. J., et al., Mass spectrometry identifies and quantifies 74 unique histone H4 isoforms in differentiating human embryonic stem cells. Proceedings of the National Academy of Sciences 105, 4093–4098 (2008)
Seligson, D. B., et al. Global histone modification patterns predict risk of prostate cancer recurrence. Nature 435, 1262–1266 (2005)
Waddington, C. H. Organisers and Genes (Cambridge, UK, Cambridge University Press, 1940)
Weber, M., et al. Chromosome-wide and promoter-specific analyses identify sites of differential DNA methylation in normal and transformed human cells. Nature Genetics 37, 853–862 (2005) doi:10.1038/ng1598 (link to article)










