


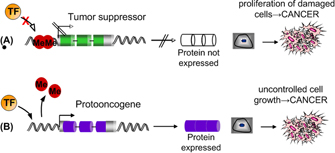
 Figure 1: Two possible mechanisms by which epigenetic modification can lead to cancer.
Figure 1: Two possible mechanisms by which epigenetic modification can lead to cancer.
















« Prev Next »
For over 50 years, we have known the structure of the DNA molecule, thanks to James Watson, Francis Crick, and the often-overlooked Rosalind Franklin (Watson & Crick, 1953). More recently, the sequencing phase of the Human Genome Project was completed in 2004 (International Human Genome Sequencing Consortium, 2004). Since then, the contents of our genome—in other words, the genes "spelled out" by the sequence, the variations in this sequence that differentiate individuals, and various other features at the DNA level—have been extensively annotated and described (International HapMap Consortium, 2003; Feuk et al., 2006; Hurles et al., 2008). The function of many human genes remains unknown, however, especially genes that do not encode proteins (Pennisi, 2007).
This wealth of genomic knowledge begs an important question: Why don't scientists better understand how genes contribute to common, complex human diseases such as cancer, heart disease, and diabetes? Environmental risk factors certainly play a significant role in these conditions, but genetic predisposition is also a causal player. Merely studying the DNA sequence has, however, produced more questions than answers, causing researchers to broaden their focus to ask what controls how and when genes are expressed. The emerging field of epigenetics (literally, "above genetics") seeks to answer such queries by focusing on nongenetically inherited changes that alter gene expression independent of the DNA sequence itself (Allis, 2007).
Methylation
One important source of epigenetic modification in humans and many other species is DNA methylation, or the addition of a methyl (-CH3 ) group to the cytosine of a CG pair in a sequence of DNA. Although the mechanisms of methylation are not entirely understood, the methyl group is usually thought to prevent transcription by recruiting protein complexes that either condense chromatin or act as a physical barrier against assembly of the transcription complex (Bird & Wolffe, 1999). The ability to alter gene expression via DNA methylation is key to maintaining the specificity and identity of different cell types in the body. After all, if cells were not selective in their gene products, this would result in a chaos of proteins and an enormous waste of cellular resources.
Imprinted versus De Novo Methylation
Some patterns of DNA methylation are inherited, or imprinted, from an organism's parents and maintained during the process of DNA replication. The growth factor IGF2 is one example of an imprinted gene, in which only the copy inherited from the father is expressed while the maternal copy is silenced (Chao & D'Amore, 2008). In contrast, de novo methylation occurs when new methylation patterns arise at specific sites in the genome as cells divide and age (Brown, 2002). Such epigenetic modifications may be due to a mutation at the DNA level (for example, a point mutation that changes a CG pair to GG), or they may develop in response to environmental factors such as diet, toxins, or smoking (Feinberg, 2007; Leng et al., 2008). The phenomenon of epigenetic change over time was recently demonstrated in a study of monozygotic (genetically identical) twins. By quantifying genome-wide methylation in 40 twin pairs, Fraga et al. (2005) found that epigenetic differences between older twin pairs were significantly greater than those between younger twins.
Methylation and Disease
Aberrant patterns of de novo methylation can lead to human disease by altering which genes are expressed in a given cell type at a given time. For example, changes in the epigenome have recently been implicated in several types of cancer (Issa, 2007). Imagine a silenced proto-oncogene losing its methylation status or, conversely, a previously unmethylated tumor suppressor gene being silenced (Figure 1). Either scenario could lead to the development of cancer.
In addition to cancer, researchers are also asking what role DNA methylation plays in other age-related complex diseases, such as heart disease. Accounting for approximately 29% of all deaths in the United States each year (Centers for Disease Control and Prevention, 2007), heart disease often manifests as a heart attack or myocardial infarction (MI). Heart attacks occur when atherosclerotic plaque that has accumulated in a coronary artery surrounding the heart ruptures and is released into the lumen of the artery. The resulting clot blocks blood flow to the heart, causing infarction. One step in plaque formation occurs when smooth muscle cells (SMCs) start to migrate away from their home in the medial layer of the artery and out toward the cell layer surrounding the lumen (Figure 2; Watkins & Farrall, 2006). But what underlies the switch in these SMCs from a quiescent into a proliferative and migratory state? Researchers hypothesize that changes in DNA methylation patterns may be at least partly responsible (Dong et al., 2002). The follow-up question, then, involves where in the genome these changes are occurring and which genes are involved. Finding these differentially methylated genes could lead to increased understanding of the biology of heart disease in particular and complex disease in general, in addition to yielding potential targets for drug intervention.
To test the hypothesis that aberrant DNA methylation patterns in SMCs cause plaque formation, researchers need an unbiased way to look for changes in methylation patterns across the genome in diseased versus nondiseased SMCs. Many methods are available, including methylated DNA immunoprecipitation, or MeDIP (Wilson, 2006), as well as methylation-specific oligonucleotide (MSO) microarrays (Gitan et al., 2002). Another emerging experimental technique is comparative methylation hybridization (CMH), which combines the same genomic clone-based approach used in the Human Genome Project with microarray technology to assay methylation status across the genome (Connelly et al., 2008). A microarray is a set of genomic sequences arranged, or arrayed, on a slide, with each spot on the array representing a known section of the human genome. For this CMH experiment, coverage of the entire human genome is needed, because it is not known which regions or genes might underlie the change in SMCs that lead to plaque formation and heart disease.
Genomic Microarrays
Although there are many types of genomic microarrays, representational oligonucleotide microarray analysis (ROMA) and NimbleGen high-density arrays, which use short DNA sequences ranging from 50 to 100 nucleotides in length to represent features across the genome, are best suited for high-density methylome studies (Feuk et al., 2006). A more cost-effective initial screening step may involve use of arrays derived from larger pieces of DNA.
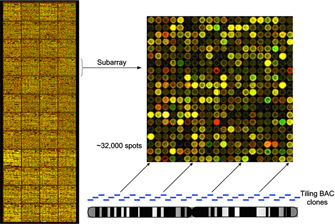
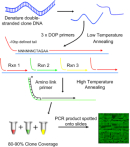
The genomic microarray used for this experiment was built from bacterial artificial chromosome (BAC) clones also used in the sequencing of the human genome. These BAC clones are composed of 100- to 150-kilobase pieces of the human genome that can be ordered along each chromosome in a tiled fashion with 50-kilobase overlapping ends (Figure 3). It takes approximately 32,000 100-kilobase BAC clones to cover the euchromatic human genome in what is called a tiling genomic array (Fiegler et al., 2003). Each spot on the array contains many copies of the BAC clone to detect differences in the test DNA samples. To reduce the complexity of the DNA sequence for hybridization, the clones are copied using a series of degenerate oligonucleotide-primed polymerase chain reactions (DOP-PCRs) (Figure 4). Each DOP-PCR primer is flanked by known 5′ and 3′ anchor sequences with six random nucleotides inserted between them, thus ensuring that the entire clone sequence will be amplified. The 5′ anchor sequence further serves to flag the amplified segments for an additional PCR step in which an amino (-NH3 ) group is linked to the end and used to affix the clone segment onto the microarray slide (Fiegler et al., 2003). To prevent false positive hybridization of repetitive DNA elements present in both the clone and the test DNA, repetitive Cot-1 DNA is first hybridized to the array and also added to the test DNA samples. Cot-1 DNA is the highly repetitive fraction of the human genome that, in the single-stranded form, is known to quickly "find" and reassociate with a complementary sequence elsewhere in the genome (Strachan & Read, 1999).
Once the microarrays are ready, the next step in CMH is to prepare the test DNA samples that will be hybridized to the array. In order to maintain cell growth and division, cells are passaged in vitro; the more passages a cell undergoes, the more times it has divided, and the "older" it is. Passaging SMCs from the aorta (aoSMCs) in culture results in cells with characteristics similar to migratory SMCs (Ying et al., 2000). Furthermore, several genes in these cells exhibit altered methylation statuses in early versus late passages (Kim et al., 2007; Post et al., 1999). These changes are also seen in atherosclerotic lesions, which suggests that epigenetic changes in proliferating SMCs could indeed be involved in the initiation and progression of heart disease. For this experiment, passage 5 (p5) aoSMCs will serve as the control, and passage 8 (p8) aoSMCs will be used as the disease sample, representing aoSMCs more prone to proliferation and migration as compared to p5 cells. Identifying differences in methylation patterns between p5 and p8 aoSMCs would support the hypothesis that changes in the epigenome contribute to myocardial infarction.
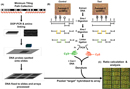
To flag methylated sites across the genome in both sets of aoSMCs, CMH uses restriction enzyme biology followed by ligation-mediated PCR. First, the DNA is treated with the restriction enzyme Sma I, which recognizes and cuts the DNA sequence CCC^GGG (Figure 5B, part i). The presence of a methyl group at the CG residue will prevent Sma I from cutting at that site. The DNA is then subjected to a second digestion by Xma I (Figure 5B, part ii), which recognizes C^CCGGG and is undeterred by a methylated CG (Inazawa et al., 2004). The resulting test DNA samples are cut into pieces, the ends of which are methylated sites with five base pairs of "sticky overhang" (-CCGGG). A linker sequence is next ligated to the sticky ends of the methylated DNA (Figure 5B, part iii). This product is then amplified using PCR so that each digested segment will be detectable on the array. PCR for the control sample, p5 aoSMCs, uses a nucleotide mixture in which the cytosines are labeled with the green fluorescent tag Cy3. On the other hand, the disease sample, p8 aoSMCs, undergoes PCR with red Cy5-labeled cytosine residues (Figure 5B, part iv). These different fluorophores are what will allow the identification of control versus disease DNA.
The control and test DNA samples are then both hybridized to the microarray slide and scanned with a microarray reader (Figure 5C). The next challenge is to translate the red and green spots into a meaningful set of results that will inform researchers of what role epigenetic changes might play in causing heart attacks. This analysis consists of comparing the relative hybridization of the Cy3-labeled control DNA (p5 aoSMCs) to the Cy5-labeled test, or disease, DNA (p8 aoSMCs). Any spot on the array in which the intensity of the green fluorescent tag Cy3 is significantly greater than the fluorescence of the red Cy5 tag represents a clone in which the control DNA is more methylated (hypermethylated) compared to the disease DNA. Conversely, any spot in which the red fluorescent signal is greater indicates a clone segment that is hypermethylated in the disease DNA and less methylated (hypomethylated) in the control. The researchers must decide how different (what level of significance) the fluorescent signals need to be to warrant further investigation, a process that involves subtracting out the noisy background and false signals that accompany most microarray experiments. The final product is a list of DNA segments differentially methylated in p5 aoSMCs versus p8 aoSMCs.
Epigenomic Profiling
So, what comes next? One hundred thousand base pairs of DNA is a vast genetic landscape, comprising anywhere from one to many genes, and it is no minor challenge to identify exactly which gene or genes are changing in methylation status. Researchers thus turn to bioinformatics resources such as genetic databases to search for likely candidate genes within the regions identified by a microarray, usually by identifying Sma I sites in the clone and finding the genes associated with those sites. Alternative lines of evidence, such as gene expression experiments and known candidates from the literature, may also be incorporated. For example, if one clone segment showing differential methylation contains a gene involved in regulating the cell cycle (i.e., cyclin-dependent kinase) and was found to have different expression levels in the p5 versus p8 samples, that region may be of particular interest for follow-up studies. The hypothesis moving forward might be that an aoSMC begins to proliferate and form plaques due to a change in methylation status of that cell's cell cycle control gene. Further experiments might include CMH using a microarray of smaller clones in selected regions to provide a higher resolution, or perhaps a single nucleotide polymorphism (SNP) array that has the power to detect methylation changes at a single nucleotide (Suzuki & Bird, 2008).
Epigenomic profiling holds great promise for understanding, predicting, and treating numerous human diseases, especially in its ability to account for the influence of environmental factors on disease states (Feinberg, 2007). Several trials of methylation-altering cancer drugs are already underway, and the potential to epigenetically treat other diseases is the next great frontier in this "post-genome" age. These discoveries will rely on successful experimental methods such as CMH to study genome-wide methylation patterns. As with many experiments, CMH yields answers and questions in roughly equal proportion. However, increased knowledge of how methylation changes in disease states informs the next set of experimental questions, thereby leading researchers closer to the ultimate goal of unlocking the epigenome.
Acknowledgements
The author acknowledges Drs. Jessica Connelly
and Simon Gregory of Duke
University's Center for
Human Genetics for generously sharing their CMH technique and editorial input. Dr.
Beth Sutton also contributed to manuscript preparation.
References and Recommended Reading
Allis, D. Epigenetics (Cold Spring Harbor, NY, Cold Spring Harbor Laboratory Press, 2007)
Bird, A. P., & Wolffe, A. P. Methylation-induced repression-Belts, braces, and chromatin. Cell 99, 451–454 (1999)
Brown, T. A. Genomes, 2nd ed. (Oxford, UK, Bios Scientific Publishers, 2002)
Centers for Disease Control and Prevention. Heart disease, www.cdc.gov/HeartDisease (2007)
Chao, W., & D'Amore, P. A. IGF2: Epigenetic regulation and role in development and disease. Cytokine and Growth Factor Reviews 19, 111–120 (2008)
Connelly, J. J., et al. Mapping DNA methylation patterns in cultured human aortic smooth muscle cells identifies epigenetic patterns that are associated with atherosclerosis [Abstract]. To be presented at the annual meeting of the American Society of Human Genetics, November 11–15, 2008, Philadelphia, PA. Available after September 25, 2008, at http://www.ashg.org/genetics/ashg08s/index.shtml (2008)
Dong, C., et al. DNA methylation and atherosclerosis. Journal of Nutrition 132, 2406S–2409S (2002)
Feinberg, A. P. Phenotypic plasticity and the epigenetics of human disease. Nature 447, 433–440 (2007) (link to article)
Feuk, L., et al. Structural variation in the human genome. Nature Reviews Genetics 7, 85–97 (2006) (link to article)
Fiegler, H., et al. DNA microarrays for comparative genomic hybridization based on DOP-PCR amplification of BAC and PAC clones. Genes, Chromosomes and Cancer 36, 361–374 (2003)
Fraga, M. F., et al. Epigenetic differences arise during the lifetime of monozygotic twins. Proceedings of the National Academy of Sciences 102, 10604–10609 (2005)
Gitan, R. S., et al. Methylation-specific oligonucleotide microarray: A new potential for high-throughput methylation analysis. Genome Research 12, 158–164 (2002)
Hurles, M. E., et al. The functional impact of structural variation in humans. Trends in Genetics 24, 238–245 (2008)
Inazawa, J., et al. Comparative genomic hybridization (CGH) arrays pave the way for identification of novel cancer-related genes. Cancer Science 95, 559–563 (2004)
International HapMap Consortium. The International HapMap Project. Nature 426, 789–796 (2003) (link to article)
International Human Genome Sequencing Consortium. Finishing the euchromatic sequence of the human genome. Nature 431, 931–945 (2004) (link to article)
Issa, J. P. Ghost in your genes [interview transcript]. NOVA, www.pbs.org/wgbh/nova/genes/issa.html (2007)
Kim, J. et al. Epigenetic changes in estrogen receptor beta gene in atherosclerotic cardiovascular tissues and in-vitro vascular senescence. Biochimica et Biophysica Acta-Molecular Basis of Disease 1772, 72–80 (2007)
Leng, S., et al. Double-strand break damage and associated DNA repair genes predispose smokers to gene methylation. Cancer Research 68, 3049–3056 (2008)
Pennisi, E. Working the (gene count) numbers—Finally, a firm answer. Science 316, 5828 (2007)
Post, W., et al. Methylation of the estrogen receptor gene is associated with aging and atherosclerosis in the cardiovascular system. Cardiovascular Research 43, 985–991 (1999)
Strachan, T., & Read, A. P. Human Molecular Genetics, 2nd ed. (Oxford, UK, Bios Scientific Publishers, 1999)
Suzuki, M. M., & Bird, A. DNA methylation landscapes: Provocative insights from epigenomics. Nature Reviews Genetics 9, 465–476 (2008) (link to article)
Watkins, H., & Farrall, M. Genetic susceptibility to coronary artery disease: From promise to progress. Nature Reviews Genetics 7, 163–173 (2006) (link to article)
Watson, J. D., & Crick, F. H. Molecular structure of nucleic acids; a structure for deoxyribose nucleic acid. Nature 171, 737–738 (1953) (link to article)
Wilson, I. M., et al. Epigenomics: Mapping the methylome. Cell Cycle 5, 155–158 (2006)
Ying, A. K., et al. Methylation of the estrogen receptor-alpha gene promoter is selectively increased in proliferating human aortic smooth muscle cells. Cardiovascular Research 46, 172–179 (2000)










