


 Figure 1: Total amount of human sequence in the High-Throughput Genomic Sequences (HTGS) division of GenBank.
Figure 1: Total amount of human sequence in the High-Throughput Genomic Sequences (HTGS) division of GenBank.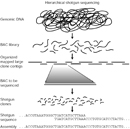
















« Prev Next »
The Human Genome Project was a 13-year-long, publicly funded project initiated in 1990 with the objective of determining the DNA sequence of the entire euchromatic human genome within 15 years. In its early days, the Human Genome Project was met with skepticism by many people, including scientists and nonscientists alike. One prominent question was whether the huge cost of the project would outweigh the potential benefits. Today, however, the overwhelming success of the Human Genome Project is readily apparent. Not only did the completion of this project usher in a new era in medicine, but it also led to significant advances in the types of technology used to sequence DNA.
Initial Principles and Goals of the Human Genome Project
From its inception, the Human Genome Project revolved around two key principles (International Human Genome Sequencing Consortium, 2001). First, it welcomed collaborators from any nation in an effort to move beyond borders, to establish an all-inclusive effort aimed at understanding our shared molecular heritage, and to benefit from diverse approaches. The group of publicly funded researchers that eventually assembled was known as International Human Genome Sequencing Consortium (IHGSC). Second, this project required that all human genome sequence information be freely and publicly available within 24 hours of its assembly. This founding principle ensured unrestricted access for scientists in academia and in industry, and it provided the means for rapid and novel discoveries by researchers of all types. At any given time, approximately 200 labs in the United States were funded by either the National Institutes of Health or the U.S. Department of Energy to support these efforts. In addition, more than 18 different countries from across the globe had contributed to the Human Genome Project by the time of its completion.
Just as the Human Genome Project revolved around two key principles, it also started with two early goals: (1) building genetic and physical maps of the human and mouse genomes, and (2) sequencing the smaller yeast and worm genomes as a test run for sequencing the larger, more complex human genome (IHGSC, 2001). When the yeast and worm efforts proved successful, the sequencing of the human genome proceeded with full force.
Phases of the Human Genome Project
© 2001 Nature Publishing Group International Human Genome Sequencing Consortium. Initial sequencing and analysis of the human genome. Nature 409, 867 (2001). All rights reserved. 
The shotgun phase of the Human Genome Project itself consisted of three
steps:
- Obtaining a DNA clone to sequence
- Sequencing the DNA clone
- Assembling sequence data from multiple clones to determine overlap and establish a contiguous sequence
The approach used by the members of the IHGSC was called the hierarchical
shotgun method, because the team members systematically generated overlapping
clones mapped to individual human chromosomes, which were individually
sequenced using a shotgun approach (Figure 2). The clones were
derived from DNA libraries made by ligating DNA fragments generated by partial
restriction enzyme digestion of genomic DNA from anonymous human donors into
bacterial artificial chromosome vectors, which could be propagated in bacteria.
When possible, the DNA fragments within the library vectors were mapped to chromosomal regions by screening for sequence-tagged sites (STSs), which are DNA fragments, usually less than 500 base pairs in length, of known sequence and chromosomal location that can be amplified using polymerase chain reaction (PCR). Library clones were also digested with the restriction enzyme HindIII, and the sizes of the resulting DNA fragments were determined using agarose gel electrophoresis. Each library clone exhibited a DNA fragment "fingerprint," which could be compared to that of all other library clones in order to identify overlapping clones. Fluorescence in situ hybridization (FISH) was also used to map library clones to specific chromosomal regions. Collectively, the STS, DNA fingerprint, and FISH data allowed the IHGSC to generate contigs, which consisted of multiple overlapping bacterial artificial chromosome (BAC) library clones spanning each of the 24 different human chromosomes (i.e., 22 autosomes and the X and Y chromosomes).
Next, individual BAC clones selected for DNA sequence analysis were further fragmented, and the smaller genomic DNA fragments were subcloned into vectors to generate a BAC-derived shotgun library. The inserts were sequenced using primers matching the vector sequence flanking the genomic DNA insert, and overlapping shotgun clones were used to generate a DNA sequence spanning the entire BAC clone. A summary of this step is shown in Figure 3. The members of the IHGSC agreed that each center would obtain an average of fourfold sequence coverage, with no clone having less than threefold coverage. The term "shotgun" comes from the fact that the original BAC clone was randomly fragmented and sequenced, and the raw DNA sequence data was then subjected to computational analyses to generate an ordered set of DNA sequences that spanned the BAC clone.

Figure 3: Levels of clone and sequence coverage.
Minimally overlapping clones are picked from a fingerprint clone contig for sequencing. The clones are sequenced to at least draft coverage to form a sequenced-clone contig. The sequences are then merged and ordered to create a sequence-contig scaffold.
© 2001 Nature Publishing Group International Human Genome Sequencing Consoritum. Initial sequencing and analysis of the human genome. Nature 409, 870 (2001). All rights reserved.
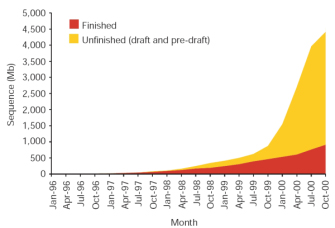
Celera: Shooting at Random and Organizing Later
Before the IHGSC had completed the first phase of the Human Genome Project, a private biotechnology company called Celera Genomics also entered the race to sequence the human genome. Led by Dr. Craig Venter, Celera proclaimed that it would sequence the entire human genome within three years. As outlined in Figure 4, Celera used two independent data sets together with two distinct computational approaches to determine the sequence of the human genome (Venter et al., 2001). The first data set was generated by Celera and consisted of 27.27 million DNA sequence reads, each with an average length of 543 base pairs, derived from five different individuals. The second data set was obtained from the publicly funded Human Genome Project and was derived from the BAC contigs (called bactigs); here, Celera "shredded" the Human Genome Project DNA sequence into 550-base-pair sequence reads representing a total of 16.05 million sequence reads. The company then used a whole-genome assembly method and a regional chromosome assembly method to sequence the human genome.
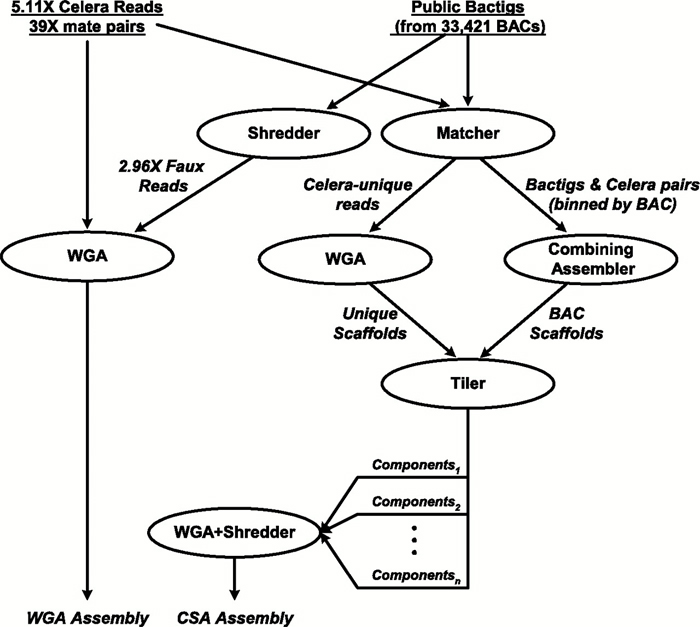
Figure 4: Architecture of Celera's two-pronged assembly strategy
Each oval denotes a computation process performing the function indicated by its label, with the labels on arcs between ovals describing the nature of the objects produced and/or consumed by a process.
© 2001 American Association for the Advancement of Science Venter, C. et al. The sequence of the human genome. Science 291, 1304–1351 (2001). All rights reserved.
In the whole-genome assembly method (also called the whole-genome random shotgun method), Celera generated a massive shotgun library derived from its own DNA sequence data combined with the "shredded" Human Genome Project DNA sequence data, which together corresponded to a total of 43.32 million sequence reads (Venter et al., 2001). Celera used computational methods and sophisticated algorithms to identify overlapping DNA sequences and to reconstruct the human genome by generating a set of scaffolds (Figure 5).
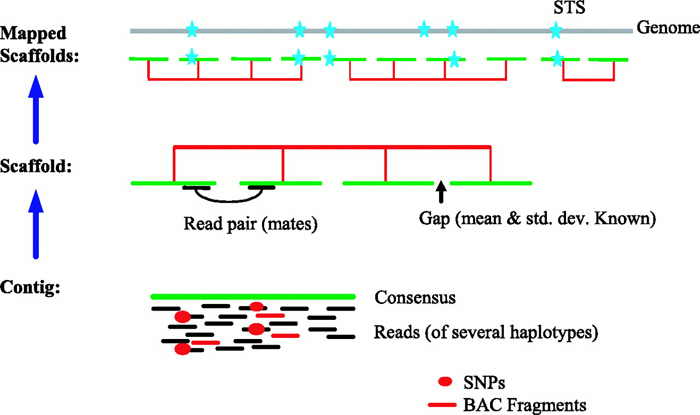
Figure 5: Anatomy of whole-genome assembly.
In whole-genome assembly, the BAC fragments (red line segments) and the reads from five individuals (black line segments) are combined to produce a contig and a consensus sequence (green line). The contigs are connected into scaffolds, shown in red, by pairing end sequences, which are also called mates. If there is a gap between consecutive contigs, it has a known size. Next, the scaffolds are mapped to the genome (gray line) using sequence tagged site (STS) information, represented by blue stars.
© 2001 American Association for the Advancement of Science Venter, C. et al. The sequence of the human genome. Science 291, 1304–1351 (2001). All rights reserved.
In contrast, with the regional chromosome assembly approach (also called the compartmentalized shotgun assembly method), Celera organized its own data and the Human Genome Project sequence data into the largest possible chromosomal segments, followed by shotgun assembly of the sequence data within each segment (Venter et al., 2001); this approach was similar to the hierarchical shotgun approach used by the IHGSC. The first step of the regional assembly approach involved separating Celera reads that matched Human Genome Project reads from those that were distinct from the public sequence data. Of the 27.27 million Celera reads, 21.38 million matched a Human Genome Project bactig, and 5.89 million did not match the public sequence data. These reads were assembled into Celera-specific or Human Genome Project-specific scaffolds, which were then combined and analyzed using whole-gene assembly algorithms. The resulting bactig data were again "shredded" to permit unbiased assembly of the combined sequence data.
Celera's whole-genome and regional chromosome assembly methods were independent of each other, permitting direct comparison of the data. Celera found that the regional chromosome assembly method was slightly more consistent than the whole-genome assembly method. Using these complementary approaches, Celera generated data that was in strong agreement with that of the IHGSC.
In February 2001, drafts of the human genome sequence were published simultaneously by both groups in two separate articles (IHGSC, 2001; Venter et al., 2001). Due to technical advances in DNA sequencing methods and a productive level of synergy between the two groups, they tied at the finish line, and both projects were completed ahead of schedule.
A Quick Lesson in DNA Sequencing
As previously mentioned, the IHGSC and Celera used different approaches to determine the sequence of the human genome. However, they used the same general method for the DNA sequencing step (Hood & Galas, 2003). This method uses DNA polymerase, the same enzyme used in DNA replication, to produce DNA sequence information. As shown in Figure 6a, DNA polymerase binds to a single-stranded DNA template and adds DNA bases to the 3′ end of the complementary DNA strand it synthesizes. DNA polymerase requires an existing primer with a free 3′ end to which it adds new DNA bases in a 5′ to 3′ manner, and it moves along the template strand in a 3′ to 5′ direction.
Researchers from both the IHGSC and Celera combined the DNA template they were interested in sequencing with DNA polymerase, a single-stranded DNA primer, free deoxynucleotide bases (dATP, dCTP, dGTP, and dTTP), and a sparse mixture of fluorescently labeled dideoxynucleotide bases (ddATP, ddCTP, ddGTP, and ddTTP) that were each labeled with a different color and would terminate new DNA strand synthesis once incorporated into the end of a growing DNA strand. The mixture was first heated to denature the template DNA strand; this was followed by a cooling step to allow the DNA primer to anneal. Following primer annealing, the polymerase synthesized a complementary DNA strand. The template would grow in length until a dideoxynucleotide base (ddNTP) was incorporated; the conditions were such that this occurred at random along the length of the newly synthesized DNA strands. In the end, the researchers were left with a mixture of newly synthesized DNA strands that differed in length by a single nucleotide, and that were labeled at their 3′ end with the color of the ddNTP-associated dye molecule (Figure 6b).
In order to determine the sequence of the newly synthesized, color-coded DNA strands, researchers needed a way to separate them based on their size, which differed by only one DNA nucleotide. To accomplish this, they electrophoresed the DNA through a gel matrix that permitted single-base differences in size to be easily distinguished. Small fragments run more quickly through the gel, and larger fragments run more slowly (Figure 6c). By putting the entire mixture into a single well of the gel, a laser can be used to scan the DNA bands as they move through the gel and determine their color; this data can be used to generate a sequence trace (also called an electropherogram), showing the color and signal intensity of each DNA band that passes through the gel (Figure 6d). The color of each band represents the final 3′ base incorporated at that position, and by reading from the bottom to the top of the gel, one can determine the sequence of the newly synthesized DNA strand from the 5′ to the 3′ end.

Figure 6: How to sequence DNA.
A) DNA polymerase binds to a single-stranded DNA template (blue) and synthesizes a complementary strand of DNA (red). B) When DNA polymerase randomly incorporates a fluorescently labeled ddNTP base, synthesis terminates. This step produces a mixture of newly synthesized DNA strands that differ in length by a single nucleotide. Each strand is labeled at the 3′ end with a fluorescently labeled ddNTP base. C) The DNA mixture is separated by electrophoresis. D) The electropherogram results show peaks representing the color and signal intensity of each DNA band. From these data, the sequence of the newly synthesized DNA strand is determined, as shown above the peaks.
© 2003 Macmillan Publishers, Ltd. Dennis, C. & Gallagher, R. (eds) The Human Genome (Palgrave, Basingstoke, 2001). Used with permission. All rights reserved.
From Rough Draft to Final Form
As stated earlier, after the completion of the draft phase of the Human
Genome Project, the IHGSC pursued the second phase of the project: the finishing
phase (IHGSC, 2004). During this phase, the researchers filled
in gaps and resolved DNA sequences in ambiguous areas that were not solved during the
shotgun phase. The finishing phase yielded 99% of the human genome in final
form. The final form of the human genome contained 2.85 billion nucleotides,
with a predicted error rate of 1 event per 100,000 bases sequenced. Furthermore,
the IHGSC reduced the number of gaps by 400-fold; only 341 gaps out of 147,821
gaps remained. The remaining gaps were associated with technically
challenging chromosomal regions. Although the earlier draft publications had
predicted as many as 40,000 protein-encoding genes, the finishing phase reduced
this estimate to between 20,000 and 25,000 protein-encoding genes. Future
challenges identified by the IHGSC during this phase included the
identification of polymorphisms as a platform for understanding genetic links to
human disease, the identification of functional elements within the genome
(genes, proteins, elements involved in gene regulation, and structural
elements), and the identification of gene and protein "modules" that act in
concert with one another.
From Digital Information to Molecular Medicine
One particularly striking finding of the Human Genome Project research is
that the human nucleotide sequence is nearly identical (99.9%) between any two
individuals. However, a single nucleotide change in a single gene can be responsible for causing human disease. Because of this, our
knowledge of the human genome sequence has also contributed immensely to our
understanding of the molecular mechanisms underlying a multitude of human
diseases. Furthermore, a merging of cytogenetic approaches with the human
genome sequence will continue to propel our understanding of human disease to
an entirely new level. Thus, although it was met with skepticism at its
inception, the Human Genome Project will certainly be heralded as one of the
most important scientific endeavors of our time.
Unfortunately, the initial hope of accelerating the discovery of new treatments for disease was not necessarily accomplished by the Human Genome Project. With the sequence of the human genome in hand, we have learned that it requires more than just knowledge of the order of the base pairs in our genome to cure human disease. Current efforts are therefore focused on understanding the protein products that are encoded by our genes. When a gene is mutated, the corresponding protein is most often defective. The emerging field of proteomics aims to understand how protein function and expression are altered in human disease states. Furthermore, investigators are also turning their attention to the expansive regions of our genome devoid of traditional protein-encoding genes. We have already started to reap the benefits of our knowledge of the human genome, and future data-mining efforts will most certainly uncover many more exciting and unexpected links to human disease.
Summary
Within a span of only 13 years, an amalgam of public and private
researchers was able to successfully complete the Human Genome Project. Although
these scientists used a number of different methods in their work, they nonetheless
obtained the same results. In doing so, the researchers not only silenced their
critics, but they also beat their own estimated project timeline by two entire
years. Perhaps even more importantly, these scientists inspired an
ongoing revolution in our fight against human disease and provided a new vision
of the future of medicine-although that future has yet to be fully realized.
References and Recommended Reading
Hood, L. & Galas, D. The digital code of DNA. Nature 421, 444–448 (2003) (link to article)
International Human Genome Sequencing Consortium. Initial sequencing and analysis of the human genome. Nature 409, 860–921 (2001) (link to article)
International Human Genome Sequencing Consortium. Finishing the euchromatic sequence of the human genome. Nature 431, 931–945 (2004) (link to article)
Venter, J. C., et al. The sequence of the human genome. Science 291, 1304–1351 (2001) (link to article)










