


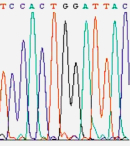
 Figure 1
Figure 1















« Prev Next »
The Human Genome Project set out to sequence the DNA of every human chromosome, thereby promising to advance knowledge of human biology and improve medicine. This project was huge in scale, as it sought to determine the order of all 3 billion nucleotides in the human genome. To reach this lofty goal, scientists developed a number of sequencing techniques that emphasized speed without too much loss of accuracy. Initially, these techniques expanded upon the so-called Sanger process that was first developed in the 1970s, gradually automating this process and increasing the number of samples that could be sequenced at one time. In fact, machines that used an automated version of Sanger method were essential to completion of various stages of the Human Genome Project. In recent years, however, researchers have increasingly begun to rely on newer and even faster methods, including the technique known as 454 sequencing.
Early DNA Sequencing Technologies
Early efforts at sequencing genes were painstaking, time consuming, and labor intensive, such as when Gilbert and Maxam reported the sequence of 24 base pairs using a method known as wandering-spot analysis (Gilbert & Maxam, 1973). Thankfully, this situation began to change during the mid-1970s, when researcher Frederick Sanger developed several faster, more efficient techniques to sequence DNA (Sanger et al., 1977). Indeed, Sanger's work in this area was so groundbreaking that it led to his receipt of the Nobel Prize in Chemistry in 1980.
Over the next several decades, technical advances automated, dramatically sped up, and further refined the Sanger sequencing process. Also called the chain-termination or dideoxy method, Sanger sequencing involves using a purified DNA polymerase enzyme to synthesize DNA chains of varying lengths. The key feature of the Sanger method's reaction mixture is the inclusion of dideoxynucleotide triphosphates (ddNTPs). These chain-terminating dideoxynucleotides lack the 3' hydroxyl (OH) group needed to form the phosphodiester bond between one nucleotide and the next during DNA strand elongation. Thus, when a dideoxynucleotide is incorporated into the growing strand, it inhibits further strand extension. The result of many of these reactions is a number of DNA fragments of varying length. These fragments are then separated by size using gel or capillary tube electrophoresis, a method in which an electric field pulls molecules across a gel substrate or hairlike capillary fiber. This procedure is sensitive enough to distinguish DNA fragments that differ in size by only a single nucleotide.
With the Sanger process, four parallel sequencing reactions are used to sequence a single sample. Each reaction involves a single-stranded template, a specific primer to start the reaction, the four standard deoxynucleotides (dATP, dGTP, dCTP, and dTTP), and DNA polymerase. The polymerase adds bases to a DNA strand that is complementary to the single-stranded sample template. One of the four dideoxynucleotides (ddATP, ddGTP, ddCTP, or ddTTP) is then added to each reaction at a lower concentration than the standard deoxynucleotides. Because the dideoxynucleotides lack the 3′ OH group, whenever they are incorporated by DNA polymerase, the growing DNA terminates. Four different ddNTPs are used such that the chain doesn't always terminate at the same nucleotide (i.e., A, G, C, or T). This produces a variety of strand lengths for analysis. Then, by putting the resulting samples through four columns on a gel (according to which dideoxynucleotide was added), researchers can see the fragments line up by size and know which base is at the end of each fragment. This makes the DNA sequence simple to read.
The Rise of Industrial Sequencing Automation
In 1986,
a company named Applied Biosystems began to manufacture automated DNA
sequencing machines based on the Sanger method (Figure 1). These machines used
fluorescent dyes to tag each nucleotide, allowing the reactions to be run in
one column and read by color (rather than by what lane they appeared in). By
running 24 samples at a time, the $100,000 machines yielded 12,000 "letters" of
DNA per day. Although the machines were more expensive than traditional glass
plates and gels, once the investment was made, these machines provided sequence
data cheaper and faster than the traditional method. In fact, these are the machines that
Craig Venter used to rally the Human Genome Project into high gear.
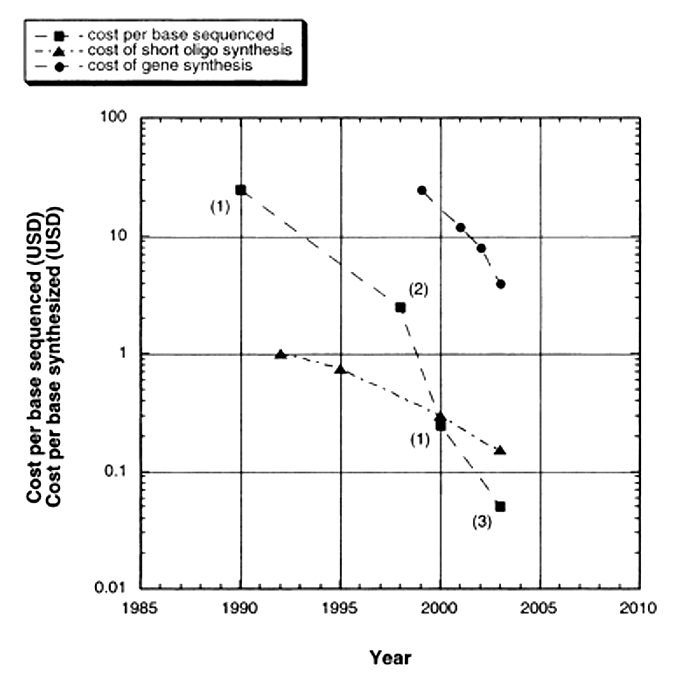
Although the basic Sanger method was still being used to sequence whole genomes, time and cost considerations continued to make it expensive, thus limiting the number of genomes that could be completed. Over time, sequencing technology (and DNA synthesis technology) advanced with more sophisticated separation strategies, alternative visualization strategies, and more parallel samples. As a result, today's machines can typically handle 96 samples at a time. In addition, whereas traditional, gel-based Sanger sequencing and early machines could only generate 250 to 500 base pairs of DNA sequence per reaction, 750 to 1,000 base pairs of sequence can now be read from a single reaction, making sequencing a much less expensive option than it used to be (Figure 2). In addition, sequencing technologies that don't use gels have been developed to further increase the efficiency of sequencing. These include flow cytometry, scanning microscopes, mass spectrometry, and hybridization strategies.
Figure 2: Cost per base of sequencing and synthesis.
A graph shows rough estimates of the cost of synthesis of DNA oligos and entire genes and raw sequencing per base. Only very limited data are available through 2003. Estimates of synthesis costs are from John Mulligan, Blue Heron Biotechnology. Historical costs of sequencing are generally not available in the literature, have not been publicized by federally funded Genome Centers, and are, in general, surprisingly hard to find.
© 2004 Mary Ann Liebert, Inc. Carlson, R. The Pace and Proliferation of Biological Technologies, Biosecurity and Bioterrorism 1, 203-214 (2004). All rights reserved. 
Draft and Complete Genomes
The Human Genome Project has issued multiple announcements about "finishing" the sequence, with landmark advances announced in 2003 and in 2006. Both of these followed a joint announcement by public (National Institutes of Health) and private (Celera) organizations that they had working drafts of a human genome in 2000. In reality, however, the complete sequencing of the human genome is still not complete because current technology has left several million bases of repeat-rich heterochromatin and many small gaps unfinished.
To better understand why this is the case, think of the human genome as an encyclopedic reading of our DNA sequence in 23 volumes. Within these volumes, there are portions of the text that differ from person to person (variability), and there are long, repetitive stretches of apparent gibberish (chapters worth of CACACA, for example). The first of these issues highlights the fact that the human genome is not a singular edition-in other words, every person on Earth has a unique genome sequence. This variability, along with the repeats within the sequence, makes it difficult for computers to assemble genomes-it's a bit like putting a together puzzle with identically shaped pieces.
Therefore, the DNA sequences announced in 2003 were actually high-quality drafts for each human chromosome. Although these sequences were informative enough to advance biomedical research, more detail was needed, because significant gaps remained in the sequence, and much of the repeat-rich heterochromatin was missing or unassembled. Then, in 2006, the DNA sequence for the 24 human chromosomes was deemed "complete." Researchers had thus filled in most of the significant remaining gaps and provided far more detail for each chromosome. Comparison of DNA sequences across populations helped with this task. Currently, scientists continue to work on perfecting the sequence, correcting the errors that occur, on average, about once every 10,000 bases.
454 Sequencing
Today, as they correct errors in the human genome sequence and work to sequence the genomes of other species, researchers are increasingly using a newer, single-nucleotide addition (SNA) method of DNA sequencing called pyrosequencing (Hyman, 1988) or 454 sequencing (after the name of the Roche-owned company that developed it). In pyrosequencing, the number of individual nucleotides is limited to the point at which DNA synthesis pauses. Then, unlike with the Sanger method, chain elongation can be resumed with the addition of nucleotides. A tiny amount of visible light is generated by enzymatic action as each nucleotide is added to a growing chain; this light is recorded as a series of peaks called a pyrogram, which corresponds to the order of the lettered nucleotides that are added and ultimately reveals the underlying DNA sequence (Metzker, 2005). Thus, by correlating when a sample flashes with the nucleotide that is present at that time, researchers can sequence a stretch of DNA (Figure 3).
In its commercial version, 454 sequencing can read up to 20 million bases per run by applying the pyrosequencing technique on picotiter plates that facilitate sequencing of large amounts of DNA at low cost compared to earlier methods. Because the method does not rely on cloning template DNA, the read is more consistent (that is, it doesn't skip unclonable segments such as heterochromatin). However, one major drawback to the pyrosequencing approach is incomplete extension through homopolymers, or simple repeats of the same nucleotide (e.g., AAAAAAA). Each read is only about 100 base pairs long at this time, making it difficult for scientists to differentiate between repeated regions longer than this length. However, pyrosequencing is improving quickly, and new machines can generate 400-base pair sequence reads.
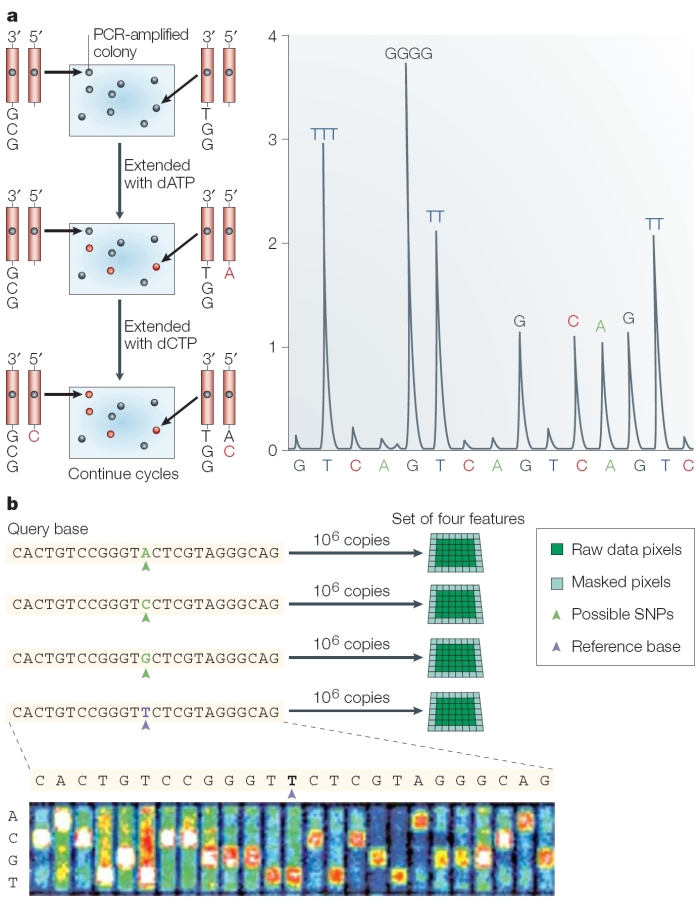
Figure 3: Examples of cyclic array sequencing and sequencing by hybridization.
a) Schematic of cyclic-array sequencing-by-synthesis methods (for example, fluorescent in situ sequencing, pyrosequencing, or single-molecule methods). Left: repeated cycles of polymerase extension with a single nucleotide at each step. The means of detecting incorporation events at individual array features varies from method to method. The sequencing templates shown here have been produced by using the POLONY method. Right: an example of raw data from Pyrosequencing, a cyclic-array method. The identity of nucleotides used at each extension step are listed along the x-axis. The y-axis depicts the measured signal at each cycle for one sequence; both single and multiple (such as homopolymeric) incorporations can be distinguished from non-incorporation events. The decoded sequence is listed along the top. b) Sequencing by hybridization. To resequence a given base, four features are present on the microarray, each identical except for a different nucleotide at the query position (the central base of 25-bp oligonucleotides). Genotyping data at each base are obtained through the differential hybridization of genomic DNA to each set of four features.
Adapted from © 2001 Cold Spring Harbor Laboratory Press. Cutler, D. J. et al. High-throughput variation detection and genotyping using microarrays. Genome Research 11, 1913–1925 (2001). All rights reserved.
Conclusion
As the previous sections illustrate, sequencing technologies both old and new have brought us information about many genomes. Beginning in the 1970s, the Sanger process made it possible for researchers to sequence stretches of DNA at speeds never before possible. Further refinement and automation of this process continued to increase sequencing rates, thereby allowing researchers to reach major milestones in the Human Genome Project well ahead of schedule. Today, newer pyrosequencing methods have drastically cut the cost of sequencing and may eventually allow every person the possibility of personalized genome information. Being able to read how our genes are expressed offers the promise of advanced medical treatments, but it will certainly require considerable work to generate, understand, organize, and apply this massive amount of data to human disease.
References and Recommended Reading
Carlson, R. The Pace and Proliferation of Biological Technologies. Biosecurity and Bioterrorism 1, 203-214 (2003) doi:10.1089/153871303769201851
Eichler, E. E., et al. An assessment of the sequence gaps: Unfinished business in a finished human genome. Nature Reviews Genetics 5, 345–354 (2004) doi:10.1038/nrg1322 (link to article)
Gilbert, W., & Maxam, A. The nucleotide sequence of the lac operator. Proceedings of the National Academy of Sciences 70, 3581–3584 (1973)
Hyman, E. D. A new method of sequencing DNA. Analytical Biochemistry 174, 423–436 (1988)
Metzker, M. L. Emerging technologies in DNA sequencing. Genome Research 15, 1767–1776 (2005)
Reinders, J., et al. Genome-wide, high-resolution DNA methylation profiling using bisulfite-mediated cytosine conversion. Genome Research 18, 469–476 (2008) doi:10.1101/gr.7073008
Sanger, F., et al. DNA sequencing with chain-terminating inhibitors. Proceedings of the National Academy of Sciences 74, 5463–5467 (1977)
Shendure, J., et al. Advanced sequencing technologies: Methods and goals. Nature Reviews Genetics 5, 335–344 (2004) doi:10.1038/nrg1325 (link to article)










