
Featured
-
-
Thesis |
 Cabinet of curiosities
Cabinet of curiosities
Michelle Francl wonders what the future will think of her office.
- Michelle Francl
-
Article |
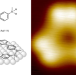 Polymerization of silanes through dehydrogenative Si–Si bond formation on metal surfaces
Polymerization of silanes through dehydrogenative Si–Si bond formation on metal surfaces
On-surface dehydrogenative bond formation between sp3-hybridized carbon atoms usually requires high temperatures. Now, it has been shown that the higher homologue, silicon, can undergo dehydrogenative polymerization at room temperature on metal surfaces. This process creates well-ordered structures on Au(111) and Cu(111), with different stereoselectivity depending on the metal.
- Lacheng Liu
- , Henning Klaasen
- & Armido Studer
-
Article |
 Stereoelectronic effects in stabilizing protein–N-glycan interactions revealed by experiment and machine learning
Stereoelectronic effects in stabilizing protein–N-glycan interactions revealed by experiment and machine learning
Analysis of the thermodynamics of protein–N-glycan interactions perturbed by mutations has revealed an enthalpy–entropy compensation that depends on the electronics of the interacting side chains. Machine-learned and statistical models showed that protein–N-glycan interactions highly correlate with stereoelectronic effects, and that a major part of protein–N-glycan interactions can be explained using the energetic rules of frontier molecular orbital interactions.
- Maziar S. Ardejani
- , Louis Noodleman
- & Jeffery W. Kelly
-
Article |
 Quasiclassical simulations based on cluster models reveal vibration-facilitated roaming in the isomerization of CO adsorbed on NaCl
Quasiclassical simulations based on cluster models reveal vibration-facilitated roaming in the isomerization of CO adsorbed on NaCl
Recent experiments reporting the isomerization of CO on a NaCl(100) surface—from C adsorbed to O adsorbed—represent a major challenge to simulate from first principles. Now, using dynamics calculations and (CO–NaCl)n cluster models that feature CO–CO interactions, it is found that isomerization occurs via a ‘roaming’ mechanism at a large distance from the NaCl(100) surface.
- Apurba Nandi
- , Peng Zhang
- & Joel M. Bowman
-
Article |
 Isolation and electronic structures of derivatized manganocene, ferrocene and cobaltocene anions
Isolation and electronic structures of derivatized manganocene, ferrocene and cobaltocene anions
Unlike ferrocene and its cationic counterpart ferrocenium, the ferrocene monoanion is an unusual species that has been observed through low-temperature electrochemical studies. Now, a family of isostructural 3d metallocenates has been isolated that consists of a manganocene, a cobaltocene and a high-spin ferrocene anion stabilized by cyclopentadienyl ligands bearing bulky aliphatic groups.
- Conrad A. P. Goodwin
- , Marcus J. Giansiracusa
- & David P. Mills
-
Article |
 Substantial π-aromaticity in the anionic heavy-metal cluster [Th@Bi12]4−
Substantial π-aromaticity in the anionic heavy-metal cluster [Th@Bi12]4−
A wide variety of organic and inorganic compounds show π-aromaticity, yet for all-metal systems it has remained restricted to compounds with three to five atoms. Now, the anionic cluster [Th@Bi12]4− has been shown to exhibit π-aromaticity, with a significant ring current despite relying on the delocalization of only two π-electrons.
- Armin R. Eulenstein
- , Yannick J. Franzke
- & Stefanie Dehnen
-
Article |
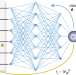 Deep-neural-network solution of the electronic Schrödinger equation
Deep-neural-network solution of the electronic Schrödinger equation
High-accuracy quantum chemistry methods struggle with a combinatorial explosion of Slater determinants in larger molecular systems, but now a method has been developed that learns electronic wavefunctions with deep neural networks and reaches high accuracy with only a few determinants. The method is applicable to realistic chemical processes such as the automerization of cyclobutadiene.
- Jan Hermann
- , Zeno Schätzle
- & Frank Noé
-
Article |
 Quantum machine learning using atom-in-molecule-based fragments selected on the fly
Quantum machine learning using atom-in-molecule-based fragments selected on the fly
Quantum machine learning with improved data efficiency and transferability has been achieved using on-the-fly selection of query-dependent training molecules, which are drawn from a ‘dictionary’ of atom-in-molecule-based fragments. The benefits of the resulting models have been demonstrated for important molecular properties and for systems including organic molecules, 2D materials, water clusters, DNA base pairs and ubiquitin.
- Bing Huang
- & O. Anatole von Lilienfeld
-
Article |
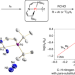 A platinum(ii) metallonitrene with a triplet ground state
A platinum(ii) metallonitrene with a triplet ground state
Transient metallonitrenes (M–N) have been proposed as key intermediates in nitrogen atom transfer reactions, but well-defined examples have remained elusive. Now, a platinum complex with an atomic nitrogen ligand, best described as a subvalent nitrogen diradical singly bonded to a platinum(ii) ion (Pt–N), has been isolated and shows ambiphilic reactivity.
- Jian Sun
- , Josh Abbenseth
- & Sven Schneider
-
Article |
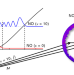 State-to-state scattering of highly vibrationally excited NO at broadly tunable energies
State-to-state scattering of highly vibrationally excited NO at broadly tunable energies
Scattering experiments in which two beams nearly co-propagate allow broadly tunable collision energies and can enable cold collisions. Now, such experiments have been combined with the preparation of NO molecules using stimulated emission to generate highly vibrationally excited states for state-to-state scattering studies, testing the theoretical gold standard in a regime not found in nature.
- Chandika Amarasinghe
- , Hongwei Li
- & Arthur G. Suits
-
Article |
 A silicon–carbonyl complex stable at room temperature
A silicon–carbonyl complex stable at room temperature
Compounds of main-group elements rarely undergo direct carbonylation reactions. Now, an electron-rich silylene intermediate has been shown to readily react with CO to form a silylene carbonyl complex that is stable at room temperature. This complex engages in CO substitution as well as oxidative addition reactions.
- Chelladurai Ganesamoorthy
- , Juliane Schoening
- & Stephan Schulz
-
News & Views |
 Delving into dynamic effects
Delving into dynamic effects
Although the application of force to induce chemical transformations is an active area of research, detailed understanding of these mechanochemical pathways is still lacking. Now, the mechanochemical activation of [4]-ladderane has been studied and found to exhibit unique non-equilibrium dynamic effects.
- Vincenzo Lordi
-
Article |
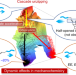 The cascade unzipping of ladderane reveals dynamic effects in mechanochemistry
The cascade unzipping of ladderane reveals dynamic effects in mechanochemistry
The mechanochemical activation of [4]-ladderane/ene has been studied and found to exhibit cascade unzipping and a consistent stereochemical distribution of products under various conditions and in different polymer backbones. Ab initio steered molecular dynamics simulations revealed unique non-equilibrium dynamic effects in the mechanochemistry of ladderane, cascade activation and reaction pathway bifurcation.
- Zhixing Chen
- , Xiaolei Zhu
- & Yan Xia
-
Article |
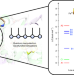 Electronic landscape of the P-cluster of nitrogenase as revealed through many-electron quantum wavefunction simulations
Electronic landscape of the P-cluster of nitrogenase as revealed through many-electron quantum wavefunction simulations
The electronic structures of the metal cofactors of nitrogenase are key to biological nitrogen fixation; however, the [Fe8S7] P-cluster and FeMo cofactor have eluded detailed electronic characterization. Now, the electronic structure of the P-cluster of nitrogenase has been revealed at the many-electron level through exhaustive quantum wavefunction simulations.
- Zhendong Li
- , Sheng Guo
- & Garnet Kin-Lic Chan
-
News & Views |
 A display of sensitivity
A display of sensitivity
Scientific progress often relies on applying published methodological advances to different problems. With the aim of improving both the uptake and reproducibility of chemical transformations, a new assessment tool has now been developed that provides a clear and easy-to-interpret overview of common factors that affect a synthetic method.
- James J. Douglas
-
Article |
 Enhanced reactivity of fluorine with para-hydrogen in cold interstellar clouds by resonance-induced quantum tunnelling
Enhanced reactivity of fluorine with para-hydrogen in cold interstellar clouds by resonance-induced quantum tunnelling
The F + para-H2 → HF + H reaction is an important source of HF in interstellar clouds; however, its unusually high rate and its dynamics at low temperature are not fully understood. Now, quantum-state resolved crossed-beam scattering measurements and anion photoelectron spectroscopy have revealed that this reactivity is caused by a resonance-enhanced tunnelling effect involving a post-barrier resonance state.
- Tiangang Yang
- , Long Huang
- & Daniel M. Neumark
-
Article |
 The photochemical ring-opening of 1,3-cyclohexadiene imaged by ultrafast electron diffraction
The photochemical ring-opening of 1,3-cyclohexadiene imaged by ultrafast electron diffraction
The photochemical electrocyclic ring-opening of 1,3-cyclohexadiene is a textbook organic chemistry reaction. Now, using ultrafast electron diffraction its reaction pathway has been resolved on the level of atomic distances and on its natural femtosecond timescale. Furthermore, coherent isomerization dynamics of the photoproduct 1,3,5-hexatriene were observed.
- T. J. A. Wolf
- , D. M. Sanchez
- & T. J. Martínez
-
Article |
 Optimization of the facet structure of transition-metal catalysts applied to the oxygen reduction reaction
Optimization of the facet structure of transition-metal catalysts applied to the oxygen reduction reaction
While much effort has been devoted to understanding how nanoparticle morphology can be leveraged to improve catalytic activity, engineering their microstructure from first principles to this end has remained difficult. Now a methodology for designing the optimal structure of a solid catalyst with the aim of achieving the highest possible activity for surface-sensitive reactions has been developed.
- M. Núñez
- , J. L. Lansford
- & D. G. Vlachos
-
Article |
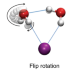 Ion-mediated hydrogen-bond rearrangement through tunnelling in the iodide–dihydrate complex
Ion-mediated hydrogen-bond rearrangement through tunnelling in the iodide–dihydrate complex
The structure and dynamics of hydrogen bonds in ion hydration shells are not yet fully understood, however, small ion–dihydrate molecular complexes represent ideal model systems with which to investigate the interplay between ion–water and water–water interactions. Now, state-of-the-art quantum dynamics simulations have provided evidence for tunnelling in hydrogen-bond rearrangements in the iodide–dihydrate complex.
- Pushp Bajaj
- , Jeremy O. Richardson
- & Francesco Paesani
-
Article |
 Relativistic quantum chemical calculations show that the uranium molecule U2 has a quadruple bond
Relativistic quantum chemical calculations show that the uranium molecule U2 has a quadruple bond
Establishing a fundamental understanding of the electronic structure of actinides remains a challenging task for both experiment and theory. Now, it is shown that for the uranium dimer, relativity and electron correlation affects not only the nature of the electronic ground state, but also lowers the bond multiplicity in comparison to previous studies.
- Stefan Knecht
- , Hans Jørgen Aa. Jensen
- & Trond Saue
-
Article |
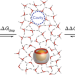 Cavitation energies can outperform dispersion interactions
Cavitation energies can outperform dispersion interactions
Binding interactions, whether between a biological receptor and ligand or between a synthetic host and guest, are frequently stronger for larger molecules than for smaller ones. This is commonly believed to arise from increased dispersion interactions, but it has now been shown that cavitation energies—always required to dissolve molecules in solution—can be more important.
- Suhang He
- , Frank Biedermann
- & Werner M. Nau
-
News & Views |
 Beyond the typical role of solvent
Beyond the typical role of solvent
State-of-the-art quantum simulations predict that solvent molecules may partner with a solute in solution to form stable chemically distinct coordination species that interconvert from one to another. The solvent would thus be directly implicated in chemical reactions.
- Gilles H. Peslherbe
-
News & Views |
 The naked truth about K+ selectivity
The naked truth about K+ selectivity
Potassium channels rapidly move K+ ions across cell membranes while blocking Na+, but how these two effects are achieved simultaneously has remained unclear. Now, extensive molecular simulations show a single mechanism that features fully dehydrated ions can explain both rapid transport and impeccable selectivity.
- Ben Corry
-
Article |
 Direct knock-on of desolvated ions governs strict ion selectivity in K+ channels
Direct knock-on of desolvated ions governs strict ion selectivity in K+ channels
That K+ channels conduct K+ ions at near-diffusion limited rates, but block the passage of smaller Na+ ions, creates an apparent contradiction. Now, atomistic simulations and free-energy calculations are used to show that both K+ permeation and ion selectivity are governed by the direct knock-on of completely desolvated ions in the channels’ selectivity filter.
- Wojciech Kopec
- , David A. Köpfer
- & Ulrich Zachariae
-
Article |
 Concerted nucleophilic aromatic substitutions
Concerted nucleophilic aromatic substitutions
Nucleophilic aromatic substitution reactions have long been thought to occur primarily via stepwise mechanisms. New and sensitive methodology for measuring carbon kinetic isotope effects now shows that most such substitutions actually occur through concerted mechanisms.
- Eugene E. Kwan
- , Yuwen Zeng
- & Eric N. Jacobsen
-
Article |
 Towards simple kinetic models of functional dynamics for a kinase subfamily
Towards simple kinetic models of functional dynamics for a kinase subfamily
Molecular dynamics simulations for seven members of the Src kinase family have now revealed a conserved step-wise deactivation process, potentially druggable intermediate states, and quantitatively similar thermodynamics and kinetics across the entire family.
- Mohammad M. Sultan
- , Gert Kiss
- & Vijay S. Pande
-
Thesis |
 Talking to Pauling’s ghost
Talking to Pauling’s ghost
Michelle Francl dusts off Pauling’s notes on bonding to explore the illusory link between electron promotion and hybridization.
- Michelle Francl
-
Article |
 Solvents can control solute molecular identity
Solvents can control solute molecular identity
Mixed quantum–classical molecular dynamics simulations of Na2 in liquid tetrahydrofuran have revealed that when local specific interactions between a solute and solvent are energetically on the same order as a hydrogen bond, the solvent controls not only bond dynamics but also the chemical identity of simple solutes.
- Devon. R. Widmer
- & Benjamin J. Schwartz
-
Article |
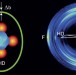 Direct observation of forward-scattering oscillations in the H+HD→H2+D reaction
Direct observation of forward-scattering oscillations in the H+HD→H2+D reaction
Our understanding of reaction dynamics has developed as more accurate measurements of product state-resolved angular distributions have become available. Now, fast forward-scattering oscillations in the product angular distribution of the benchmark chemical reaction H + HD → H2 + D have been observed and are in excellent agreement with quantum-mechanical dynamics calculations.
- Daofu Yuan
- , Shengrui Yu
- & Xueming Yang
-
Article |
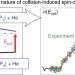 Understanding the quantum nature of low-energy C(3Pj) + He inelastic collisions
Understanding the quantum nature of low-energy C(3Pj) + He inelastic collisions
Collision-induced spin–orbit transitions involve multiple interaction potentials and are by nature non-adiabatic, complicating both their experimental and theoretical study. Crossed-beam experiments and non-Born–Oppenheimer quantum calculations for inelastic collisions of carbon atoms with helium atoms, down to energies corresponding to temperatures below 10 K, have now been performed. Quantum-dynamical resonances predicted by theory were experimentally detected.
- Astrid Bergeat
- , Simon Chefdeville
- & François Lique
-
Article |
 O2−O2 and O2−N2 collision-induced absorption mechanisms unravelled
O2−O2 and O2−N2 collision-induced absorption mechanisms unravelled
Molecular collisions can lead to the absorption of incident light even for transitions that are spectroscopically forbidden for the isolated molecules. Now the electronic–vibrational transitions of O2 have been theoretically studied and, contrary to textbook knowledge, it is shown that the absorption mechanism and the spectral line shape depend on the collision partner, oxygen or nitrogen.
- Tijs Karman
- , Mark A. J. Koenis
- & Gerrit C. Groenenboom
-
Article |
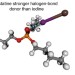 Experimental and computational evidence of halogen bonds involving astatine
Experimental and computational evidence of halogen bonds involving astatine
Halogen bonding is known to get stronger with increasing halogen polarizability, but some trends of the periodic table break down for heavy elements owing to relativistic effects. Now, through distribution coefficient measurements and relativistic quantum mechanical calculations, AtI has been shown to form stronger halogen bonds than I2—meaning that astatine conforms to the trend.
- Ning Guo
- , Rémi Maurice
- & Nicolas Galland
-
News & Views |
 Sticky when wet
Sticky when wet
The aqueous hydronium cation diffuses about twice as fast as the aqueous hydroxide anion in liquid water, but the origin of this behaviour has been unclear. Now, state-of-the-art simulations provide an explanation for this long-standing conundrum.
- Ji Chen
- & Angelos Michaelides
-
Article |
 Hydroxide diffuses slower than hydronium in water because its solvated structure inhibits correlated proton transfer
Hydroxide diffuses slower than hydronium in water because its solvated structure inhibits correlated proton transfer
Even though the Grotthuss mechanism was proposed two centuries ago, it is still unclear why proton transfer via the hydroxide ion is slower than that via hydronium. Advanced ab initio molecular dynamics simulations now show that it is because proton transfer via hydroxide is less temporally correlated than transfer via hydronium.
- Mohan Chen
- , Lixin Zheng
- & Xifan Wu
-
Article |
 Scattering resonances in bimolecular collisions between NO radicals and H2 challenge the theoretical gold standard
Scattering resonances in bimolecular collisions between NO radicals and H2 challenge the theoretical gold standard
Calculations at the theoretical gold standard generally yield accurate results for a variety of energy-transfer processes in molecular collisions. Using anti-seeding methods in a crossed-beam inelastic scattering experiment, a resonance structure is clearly resolved for NO–H2 collisions, pushing the required accuracy for theoretical potentials beyond the gold standard.
- Sjoerd N. Vogels
- , Tijs Karman
- & Sebastiaan Y. T. van de Meerakker
-
News & Views |
 Three is the magic number
Three is the magic number
Although predicted many years ago, chemically reactive termolecular reactions were thought to be unimportant in defining the behaviour of combustion systems. Now, calculations have shown that such reactions between radicals and long-lived bimolecular complexes can actually play an important role in hydrogen combustion.
- Rex T. Skodje
-
Article |
 Ephemeral collision complexes mediate chemically termolecular transformations that affect system chemistry
Ephemeral collision complexes mediate chemically termolecular transformations that affect system chemistry
Chemically termolecular reactions — arising from the collision of ephemeral collision complexes with other chemically reactive species — have been neglected in current gas-phase chemical mechanisms of combustion and planetary atmospheres. First-principles calculations reveal that such chemically termolecular reactions constitute major pathways affecting macroscopic observables.
- Michael P. Burke
- & Stephen J. Klippenstein
-
Article |
 Complete protein–protein association kinetics in atomic detail revealed by molecular dynamics simulations and Markov modelling
Complete protein–protein association kinetics in atomic detail revealed by molecular dynamics simulations and Markov modelling
Uncovering the microscopic details of protein–protein association via direct molecular dynamics (MD) simulations has been prevented by the excessive lifetimes of associated states. Now, association and dissociation for the barnase–barstar complex has been studied by adaptive high-throughput MD simulations and Markov modelling, revealing intermediate structures, energetics and kinetics on microseconds-to-hours timescales.
- Nuria Plattner
- , Stefan Doerr
- & Frank Noé
-
News & Views |
 Caught in the act
Caught in the act
Femtochemistry, the real-time study of reactions on a timescale that captures the molecular and atomic activity involved, has traditionally been performed in the gas or liquid phase. It has now been extended to the solid state in a study that highlights how a controlled reaction environment can place steric constraints on the motions of photoproducts.
- Giulio Cerullo
- & Marco Garavelli
-
Article |
 Transition-metal-free chemo- and regioselective vinylation of azaallyls
Transition-metal-free chemo- and regioselective vinylation of azaallyls
Cross coupling under transition-metal-free conditions is an attractive and economic alternative to traditional transition-metal-catalysed methods. Metal-free coupling of azaallyls has now been demonstrated with vinyl bromide electrophiles, delivering allylic amines in excellent yields. Moreover, mechanistic evidence supports dual reaction pathways triggered by azaallyl anions and radicals.
- Minyan Li
- , Osvaldo Gutierrez
- & Patrick J. Walsh
-
Article |
 Coherent ultrafast lattice-directed reaction dynamics of triiodide anion photodissociation
Coherent ultrafast lattice-directed reaction dynamics of triiodide anion photodissociation
Dissociative reactions in the solid state are prone to sample damage. Now, improved sample handling and measurement conditions enable the study of the dissociative reaction of a model triatomic system in the solid state on ultrafast timescales, revealing the significant impact of lattice coordination on the reaction pathway.
- Rui Xian
- , Gastón Corthey
- & R. J. Dwayne Miller
-
Article |
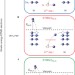 Observation of electron-transfer-mediated decay in aqueous solution
Observation of electron-transfer-mediated decay in aqueous solution
Electron-transfer-mediated decay (ETMD) is a recently discovered type of electronic relaxation that involves the refilling of a core hole by an electron from a neighbouring species. It has now been observed in LiCl solution, when previously it had only been seen in rare-gas clusters. Spectra generated during ETMD are observed to be sensitive to the immediate environment of the initially ionized ion.
- Isaak Unger
- , Robert Seidel
- & Nikolai V. Kryzhevoi
-
News & Views |
 Encoding evolution of porous solids
Encoding evolution of porous solids
The design and prediction of network topology is challenging, even when the components' principle interactions are strong. Now, frameworks with relatively weak 'chiral recognition' between organic building blocks have been synthesized and rationalized in silico — an important development in the reticular synthesis of molecular crystals.
- Caroline Mellot-Draznieks
- & Anthony K. Cheetham
-
Perspective |
 Oriented electric fields as future smart reagents in chemistry
Oriented electric fields as future smart reagents in chemistry
No longer a theoretical dream, this Perspective describes effects of oriented external electric fields on rates and selectivity patterns of nonpolar reactions. Discussions of the Diels–Alder reaction, C–H and C=C bond activations and so on, underscore the potential usage of oriented electric fields as future smart catalysts, inhibitors and reagents in chemistry.
- Sason Shaik
- , Debasish Mandal
- & Rajeev Ramanan
-
Article |
 Reticular synthesis of porous molecular 1D nanotubes and 3D networks
Reticular synthesis of porous molecular 1D nanotubes and 3D networks
Porous molecular crystals have desirable properties, but are hard to form with the level of structural control seen for extended framework materials. Now, a ‘mix-and-match’ chiral recognition strategy has been used to form reticular porous supramolecular nanotubes and 3D networks, providing a blueprint for pore design in molecular crystals.
- A. G. Slater
- , M. A. Little
- & A. I. Cooper
-
Article |
 Unexpected mechanochemical complexity in the mechanistic scenarios of disulfide bond reduction in alkaline solution
Unexpected mechanochemical complexity in the mechanistic scenarios of disulfide bond reduction in alkaline solution
The controlled mechanical activation of specific covalent bonds is a rapidly expanding field in chemistry. Now, it is shown that disulfide bond reduction proceeds through different mechanisms depending on the external force applied. This strongly suggests that refined models should be used when interpreting mechanochemical experiments, particularly when sonication is involved.
- Przemyslaw Dopieralski
- , Jordi Ribas–Arino
- & Dominik Marx
-
News & Views |
 Fixing Jacob's ladder
Fixing Jacob's ladder
Density functional theory calculations can be carried out with different levels of accuracy, forming a hierarchy that is often represented by the rungs of a ladder. Now a new method has been developed that significantly improves the accuracy of the 'third rung' when calculating the properties of diversely bonded systems.
- Roberto Car
-
Article |
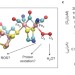 Mechanism of O2 diffusion and reduction in FeFe hydrogenases
Mechanism of O2 diffusion and reduction in FeFe hydrogenases
FeFe hydrogenases are highly efficient H2 producing enzymes; however, they can be inactivated by O2. Now, a mechanism for O2 diffusion within FeFe hydrogenases and its reactions at the active site of the enzyme has been proposed. These findings could help with the design of hydrogenase mutants with increased resistance to oxidative damage.
- Adam Kubas
- , Christophe Orain
- & Christophe Léger
-
Article |
 Engineering nanometre-scale coherence in soft matter
Engineering nanometre-scale coherence in soft matter
The existence (or not) of electronic coherence in homopolymers is dependent on a balance between monomer–monomer interactions and environmental heterogeneity. Now, by understanding how even–odd orbital symmetry influences coherence and produces resistance oscillations as a function of distance—it is shown that DNA sequences can be designed to support coherent charge transport.
- Chaoren Liu
- , Limin Xiang
- & Nongjian Tao
 SARS-CoV-2 simulations go exascale to predict dramatic spike opening and cryptic pockets across the proteome
SARS-CoV-2 simulations go exascale to predict dramatic spike opening and cryptic pockets across the proteome