
Featured
-
-
Article |
 Deep learning study of tyrosine reveals that roaming can lead to photodamage
Deep learning study of tyrosine reveals that roaming can lead to photodamage
Amino acids are one of the major building blocks of life, but the ways in which they respond to light excitation are not fully understood. Now, the photochemistry of tyrosine has been studied using physically inspired deep neural networks, leading to the observation of unconventional dynamically controlled reactivity that involves ‘roaming’ radicals that can cause photodamage.
- Julia Westermayr
- , Michael Gastegger
- & Philipp Marquetand
-
Comment |
 Best practices in machine learning for chemistry
Best practices in machine learning for chemistry
Statistical tools based on machine learning are becoming integrated into chemistry research workflows. We discuss the elements necessary to train reliable, repeatable and reproducible models, and recommend a set of guidelines for machine learning reports.
- Nongnuch Artrith
- , Keith T. Butler
- & Aron Walsh
-
Article |
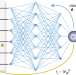 Deep-neural-network solution of the electronic Schrödinger equation
Deep-neural-network solution of the electronic Schrödinger equation
High-accuracy quantum chemistry methods struggle with a combinatorial explosion of Slater determinants in larger molecular systems, but now a method has been developed that learns electronic wavefunctions with deep neural networks and reaches high accuracy with only a few determinants. The method is applicable to realistic chemical processes such as the automerization of cyclobutadiene.
- Jan Hermann
- , Zeno Schätzle
- & Frank Noé
-
News & Views |
 A display of sensitivity
A display of sensitivity
Scientific progress often relies on applying published methodological advances to different problems. With the aim of improving both the uptake and reproducibility of chemical transformations, a new assessment tool has now been developed that provides a clear and easy-to-interpret overview of common factors that affect a synthetic method.
- James J. Douglas
-
Article |
 Optimization of the facet structure of transition-metal catalysts applied to the oxygen reduction reaction
Optimization of the facet structure of transition-metal catalysts applied to the oxygen reduction reaction
While much effort has been devoted to understanding how nanoparticle morphology can be leveraged to improve catalytic activity, engineering their microstructure from first principles to this end has remained difficult. Now a methodology for designing the optimal structure of a solid catalyst with the aim of achieving the highest possible activity for surface-sensitive reactions has been developed.
- M. Núñez
- , J. L. Lansford
- & D. G. Vlachos
-
Feature |
 Quantum reform
Quantum reform
Quantum computers potentially offer a faster way to calculate chemical properties, but the exact implications of this speed-up have only become clear over the last year. The first quantum computers are likely to enable calculations that cannot be performed classically, which might reform quantum chemistry — but we should not expect a revolution.
- Leonie Mueck
-
News & Views |
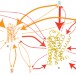 A stitch in time
A stitch in time
Lengthy molecular dynamics simulations of complex systems at the atomic scale usually require supercomputers. Now, by stitching together many shorter independent simulations run 'in the cloud', this requirement has been circumvented, allowing two milliseconds of the dynamics of a G-protein-coupled receptor to be simulated.
- Xavier Deupi
-
Article |
 Cloud-based simulations on Google Exacycle reveal ligand modulation of GPCR activation pathways
Cloud-based simulations on Google Exacycle reveal ligand modulation of GPCR activation pathways
Two milliseconds of molecular dynamics simulations of a major drug-target G-protein-coupled receptor (GPCR) has been carried out using Google's Exacycle cloud computing platform. Markov state models were used to aggregate independent simulations into a statistical model that provides an atomistic description of GPCR ligand-modulated activation pathways.
- Kai J. Kohlhoff
- , Diwakar Shukla
- & Vijay S. Pande
 Direct observation of geometric-phase interference in dynamics around a conical intersection
Direct observation of geometric-phase interference in dynamics around a conical intersection