
Abstract
Goats contribute significantly to the global food security and industry. They constitute a main supplier of meat and milk for large proportions of people in Egypt and worldwide. Brucellosis is a zoonotic infectious disease that causes a significant economic loss in animal production. A case–control genome-wide association analysis (GWAS) was conducted using the infectious status of the animal as a phenotype. The does that showed abortion during the last third period of pregnancy and which were positive to both rose bengal plate and serum tube agglutination tests, were considered as cases. Otherwise, they were considered as controls. All animals were genotyped using the Illumina 65KSNP BeadChip. Additionally, the diversity and composition of vaginal and fecal microbiota in cases and controls were investigated using PCR-amplicone sequencing of the V4 region of 16S rDNA. After applying quality control criteria, 35,818 markers and 66 does were available for the GWAS test. The GWAS revealed a significantly associated SNP (P = 5.01 × 10–7) located on Caprine chromosome 15 at 29 megabases. Four other markers surpassed the proposed threshold (P = 2.5 × 10–5). Additionally, fourteen genomic regions accounted for more than 0.1% of the variance explained by all genome windows. Corresponding markers were located within or in close vicinity to several candidate genes, such as ARRB1, RELT, ATG16L2, IGSF21, UBR4, ULK1, DCN, MAPB1, NAIP, CD26, IFIH1, NDFIP2, DOK4, MAF, IL2RB, USP18, ARID5A, ZAP70, CNTN5, PIK3AP1, DNTT, BLNK, and NHLRC3. These genes play important roles in the regulation of immune responses to the infections through several biological pathways. Similar vaginal bacterial community was observed in both cases and controls while the fecal bacterial composition and diversity differed between the groups (P < 0.05). Faeces from the control does showed a higher relative abundance of the phylum Bacteroidota compared to cases (P < 0.05), while the latter showed more Firmicutes, Spirochaetota, Planctomycetota, and Proteobacteria. On the genus level, the control does exhibited higher abundances of Rikenellaceae RC9 gut group and Christensenellaceae R-7 group (P < 0.05), while the infected does revealed higher Bacteroides, Alistipes, and Prevotellaceae UCG-003 (P < 0.05). This information increases our understanding of the genetics of the susceptibility to Brucella in goats and may be useful in breeding programs and selection schemes that aim at controlling the disease in livestock.
Similar content being viewed by others

Introduction
Brucellosis is a bacterial infectious disease caused by various Brucella species1. It is categorized as a complex disease due to its wide range of hosts and the variable clinical signs1. It is a major zoonotic disease that infects domesticated and wild animals2,3, and is classified as a serious challenge and a neglected disease in developing countries, infecting about half a million people yearly4,5. Brucellosis is endemic and a highly contagious disease in the mediterranean countries, the middle east, south and central Asia and central and south America4,6,7. The disease causes significant economic losses to the animal production industry due to reducing animal productivity and fertility, fetuses’ losses, vaccination and veterinary care and culling costs5,8,9,10.
Brucella melitensis is the most causative prevalent species of the disease in small ruminants11. Animals become infected after the ingestion of contaminated milk, feed, water or grazing forage, close contact with infected animals, uterine secretions, or aborted foetuses12,13,14. The most notable clinical signs of the disease are abortion during the last third part of pregnancy, damages in male congenital tract with sterility, reduced fertility, reduced weight gain, a substantial decline in milk production and serious clinical complications to humans as well5,15. Humans acquire the disease through direct contact with infected animals or consumption of the contaminated animal products. Different Brucella spp. may infect humans16, however, B. melitensis transmitted from sheep and goats is the most contagious virulent pathogen6,17,18. However, data for the Brucella prevalence in Egypt are scarce, recent studies stated that the disease is endemic with high prevalence19,20,21. The prevalence of brucellosis in Egyptian goats ranged from 3.5521 to 11.3% according to different resources22,23 and causes a severe reduction in the profitability of animal production18.
Currently, there is no safe and effective Brucella vaccine available for use in humans24. Vaccines are available for animals but do not differentiate between infected and vaccinated animals on serological diagnostic tests25. Other tests, including molecular methods, have been proposed for the diagnosis of brucellosis in farm animals, although a combination of different methods appears most reliable for definitive identification so far26. It is well-known that genetics contribute significantly to complex traits27, including animal health and welfare28,29. Improvement in animal health through genetic selection is advantageous, because genetic gain is accumulative and permanent, as the polymorphisms introduced by breeding into a population can persist for many generations30. Identification of the host genetic predisposition to disease susceptibility could be a useful aid in programs that focus on management, screening, and culling of diseased animals31. It is worth to note that the vaginal and fecal microbiota has direct relationships to animal performance as it affects animal fertility32, and thus the profitability of animal farms. Therefore, exploring the vaginal and fecal microbiome could assist to predict the reproduction success and may be considered in selection programs33,34.
Few genetic variants and candidate genes have been identified to contribute to brucellosis in humans35,36,37,38 and livestock39,40,41,42,43. This suggests that there are individual variations in the degree of susceptibility or resistance to the disease44. Besides, understanding the changes in the vaginal and gut microbiota in case and control animals could improve their reproductive performance32,45. Limited information is available for the disease related traits and its effect on the vaginal and gut microbiome in livestock. The objective of this study was to scan animals’ genomes to identify genetic markers and candidate genes underlying susceptibility to brucellosis infection in Egyptian goats. Also, the effect of brucellosis infection on the diversity and composition of vaginal and fecal microbial community was considered. This may help in management system and controlling programs of the disease.
Materials and methods
Population and phenotype definition
All animal procedures included in the current study were approved by the animal breeding committee at the Desert Research Center (DRC) in Egypt. Along with the relevant ARRIVE (https://arriveguidelines.org/), all methods were performed in accordance with the relevant guidelines and regulations. A total number of 96 Damascus goats from two independent flocks located in the North-costal region of Egypt, were included in this study. The most obvious sign of brucellosis infection was recorded in the first flock (n = 76). More than half of the does have aborted at the last third period of pregnancy with retained placenta, while the remaining does have delivered their kids successfully. The second flock consisted of 20 does were included in the analysis as controls because they all gave successful birth, and they were negative to all the subsequent brucellosis serological tests. All the animals received a similar diet consisting of alfalfa hay (50%) and barley grains (50%). All the samples used in this study were collected during the first week after abortion or delivery.
Blood samples for each animal in the study were collected from the jugular vein in vacutainer tubes containing EDTA and other plain tubes without anticoagulant. Subsequently, serum samples were initially tested against Brucella infection using the Rose Bengal Plate test (RPPT)46. Samples that showed positive reaction to RPPT were tested using serum tube agglutination test (STAT)47 to confirm the infection. For RPPT, equal volumes (30 μL) of standardized B. melitensis antigen and test serum were mixed thoroughly. Any appearance of agglutination was recorded as a positive result. According to the degree of agglutination, positive samples were classified as weakly positive ( +) to strongly positive (+ + + +). The samples in which agglutination was not observed within 4 min were assigned to be negative ( −) to Brucella infection. Using STAT, significant titers were those determined to be ≥ 1/80, which were considered positive to Brucella infection, while the 1/40 titers were considered negative. According to the World Organization of Animal Health protocol, RPPT and STAT tests were repeated for the same animals twice with a period of 2 weeks between the two tests to confirm the infection (case vs control). Additionally, another criterion was taken into account, which was the reproductive status of the animal (abortion vs birth).
Finally, animals were considered cases if they have aborted at the last third period of pregnancy and were positive to both RPPT and STAT tests. Otherwise, they were considered control and matched with cases on the flock and proximal birth date.
Goat genotyping
Genomic DNA was extracted from the whole blood of each doe using a Puregen Core Genomic DNA extraction from blood Kit A (Qiagen®, Hilden, Germany) according to the manufacturer’s protocol. The quantity and quality of extracted DNA was assessed using Nanodrop spectrophotometer. High-quality DNA samples (≥ 50 ng/µL) were genotyped at the Research Institute for Farm Animal Biology, Dummerstorf, Germany, using the Illumina®inc. Goat_IGGC_65K_v2 Infinium HD SNP chip (Illumina, San Diego, CA, USA), which contains 59,727 SNPs in total, evenly distributed throughout the caprine genome using iScan Reader (Illumina). The ARS1 goat assembly was used as a reference genome in this study. Genotype calling was performed using GenomeStudio software (Illumina) according to the manufacturer’s protocols.
A principal component analysis (PCA) was performed to illustrate the relationship between individuals using R software. The PC1 and PC2 (Fig. 1) that accounted for 6.7% and 4.64% of the genetic variation in the studied population, respectively, were only considered in the subsequent analysis. The genotyped population was filtered for quality in PLINK v1.9 software48, using the following parameters: (i) significant deviation from Hardy–Weinberg Equilibrium (HWE) p < 10−6; (ii) Minor Allele Frequency (MAF ≤ 0.01); (iii) genotype call rate < 0.99 for markers and < 90% for individuals. Furthermore, SNPs with unknown or identical chromosomal positions were also excluded from the subsequent analyses. A relatedness test was done using PLINK v1.9 to verify independence among the individuals. To avoid genomic inflation (i.e. the cryptic population substructure caused by the presence of closely related animals in the absence of pedigree information), pairwise identity-by-decent (IBD) was estimated for each pair of individuals in the population. Individuals of a pair that had a pi-hat value greater than 0.45 were considered as closely related, and thus were removed from the analysis.

A Principal component analysis (PCA) plot representing the genetic landscape of 10 horse breeds extended across first and second components (PC1 and PC2) derived from eigen vectors and eigen values obtained from eigen decomposition of a genotypic (co)variance matrix between all individuals. Each color shows a different breed, and each point represents 1 sample.
Genome-wide association analysis
Two statistical approaches based on fixation index (Fst) and genome-wide association analysis were implemented in SNPRelate package in R49 and with the –assoc function in PLINK software v1.9, respectively. The Bonferroni correction of P-values was applied by accounting for the number of performed tests, suggesting a significance threshold of P < 5 × 10–6. To avoid false negative SNPs, another threshold of P < 5 × 10–5 was used following the Welcome Trust Case Control Consortium suggestion (WTCCC)50, which was also suggested by Duggal P. et al.51 as a conservative way to correct for the SNPs that are not truly independent. A quantile–quantile (Q-Q) plot of the observed P-values against expected P-values was created to evaluate the overall GWAS associations. A Manhattan plot of the negative common logarithm of SNP-specific P-values versus the chromosomal location was drawn using qqman package in R52.
Variance components and heritability estimate
Due to the absence of pedigree data for animals included in this study, variance was proportioned, and SNP-based heritability (h2) was estimated based on the genomic information only using the Genome-wide complex trait analysis (GCTA) software53. First, restricted maximum likelihood (REML) analysis was performed until convergence of the likelihood ratio test (LRT). This option was followed by the option ‘–grm’ to estimate the variance explained by the SNPs that were used to estimate the genomic relationship matrix (GRM). Second, random effects were predicted by the best linear unbiased prediction (BLUP) method using ‘–reml-pred-rand’ to estimate the breeding value for each animal in this study attributed by the aggregative effect of the SNPs used to estimate the GRM followed by ‘–blup-snp’ flags implemented in GRMEL tool, which calculates the BLUP solutions for the SNP effects.
Genetic variance explained by markers
The single-step GBLUP implemented in the BLUPF90 family54 was used to estimate the SNP effects from genomic estimated breeding values (GEBVs) of genotyped animals using the postGSf90 software of the BLUPF90 package55. SNP effects were calculated as: û = DZ' [ZDZ'] –1 âg, where û is the vector of SNP effect; D is the diagonal matrix for weighting factors of the SNP effect; Z is the matrix of genotypes, and âg is the vector of breeding values predicted for genotyped animals.The variance explained by each SNP was calculated as: σ2 = û2 2p(1 − p), where û is the SNP effect described above and p is the allele frequency of the SNP56. The percentage of genetic variance explained by a window segment of 5 adjacent SNPs was calculated as: (Var (ai))/(σ2a) × 100%, where ai is the genetic value of the i-th region that consists of 5 adjacent SNP and σ2a is the total genetic variance57.
Functional annotation of the significant SNPs
For each significant SNP (SNPs with a P-value equal to or exceeding the genome-wide threshold (P-value < 5 × 10–5) and the top 10 SNPs that accounted for the highest Fst values, a defined 2 Mb region (1 Mb on each side) was considered as a QTL interval. SNP locations reported in this paper are based on the genome version of Capra hircus available from the National Center for Biotechnology Information (ARS1, NCBI). Information on the SNPs and functional annotation of genes were obtained from BioMart at the Ensembl Genome Browser (http://www.ensembl.org/biomart)58. Functions of genes and encoded proteins were investigated using UniProt, OMIA (Online Mendelian Inheritance in Animals) and the GeneCards databases59.
Vaginal and fecal microbiota
Based on the infection definition mentioned above, fecal and vaginal swab samples were collected from case and control animals included in the study (n = 8, each).
The vaginal samples were collected from each animal independently by inserting a sterile cotton swab into the vagina and rolled on the surface of vaginal epithelium for 30 s. The collected swabs were then stored at − 20 °C until subsequent analyses. The frozen swabs were thawed, re-suspended in 5 ml dissociation solution (0.1% Tween 80, 1% methanol, and 1% tertiary butanol (vol/vol), pH = 2), vortexed for 1 min and the supernatant solution was subsequently transferred to another sterile 50 ml tube. This step was further repeated twice and supernatants were collected. Then, the liquid was centrifuged at 12,000×g for 20 min to collect cell pellets that were used in DNA isolation using QIAamp DNA Stool Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions.
Fecal samples were collected from the rectum of the goats using clean gloves and samples were placed in sterilized 50 ml tubes that were frozen immediately at − 20 °C. Subsequently, 0.2 g fecal samples were used in DNA extraction using QIAamp DNA Stool Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions.
PCR amplification, library sequencing, and data analysis
The quality and quantity of extracted nucleic acid were checked by Nanodrop spectrophotometer. The composition and diversity of microbial communities were studied by amplification of variable region V4 of the 16S rDNA gene by 515F and 926R primer sets using the following PCR conditions: 94 °C for 3 min; 35 cycles of 94 °C for 45 s, 50 °C for 60 s, and 72 °C for 90 s; and 72 °C for 10 min. The purified PCR-amplicons were sequenced using the Illumina MiSeq system at Integrated Microbiome Resource (IMR, Dalhousie University, Halifax, NS, Canada).
The generated paired-end (PE) Illumina raw sequences were analyzed in R software using the DADA2 pipeline60. The Fastq files of paired-end reads were demultiplexed and their quality was checked. Then after, the sequences were filtered, trimmed, and dereplicated followed by merging R1 and R2 reads together to get denoised sequences. The denoised sequences were subjected to removing the chimeras; then Amplicon Sequence Variants (ASVs) were obtained. Taxonomic assignment of ASVs was conducted using “assign Taxonomy” and “addSpecies” functions and microbial taxa were identified using SILVA reference database (version 138). Alpha diversity indices, including Chao1, Shannon, and InvSimpson were calculated. Moreover, beta diversity of microbial communities was calculated as principal coordinate analysis (PCoA) using bray–curtis dissimilarity. The differences in diversity indices and the relative abundances of bacterial phyla and genera were estimated by unpaired T-test. The raw sequence reads were deposited to SRA at https://www.ncbi.nlm.nih.gov/sra/PRJNA910086.
Results
The quality control filtration process removed 15,051 SNPs and 10 animals, while 44,353 variants and 66 does passed QC criteria. The remaining SNPs were pruned for linkage disequilibrium using a window size of 1 Mb and a threshold of r2 > 0.5. Finally, 66 individuals (12 cases and 54 controls) genotyped at 35,818 SNPs were used in the subsequent GWAS analyses.
Case–control GWAS
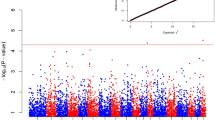
Plotting the corrected P-values for most SNPs in the GWAS analysis exhibited a good correspondence to the expected P-values under the null hypothesis of no association, with the available number of SNPs indicating association with the trait under study (Fig. 2). The SNP array scans for brucellosis infection in Damascus goats (Fig. 3A) revealed that a SNP (snp1723-scaffold1048-1212160; P-value < 5.0 × 10–7) on chromosome 15 at 29 Mb surpassed the suggested genome-wide significance threshold level. The top 10 SNPs, some of which reach the WTCCC suggested threshold were located on chromosomes 17 (25 Mb), 5 (21 Mb), 20 (9 Mb), 3 (54 Mb), 2 (102 Mb), 12 (30 Mb), 18 (7 Mb), 5 (75 Mb) and 30 (39 Mb) (Table 1). The full details of the association test of all SNPs, MAF and SNP effect are located in the supplementary excel file. Among those, 3 SNPs were located within genes on chromosome 2 (102 Mb and 21 Mb) and chromosome 20 (9 Mb), while the remaining 7 SNPs were located in intergenic regions. Some of the significantly associated chromosomal regions harbored coding genes with biological roles contributing to T-cell signaling and controlling of the immune regulations and responses to infections.

The Q-Q plot of the genome-wide association, where the -log10-transformed observed P-values (y-axis) are plotted against -log10-transformed expected P-values (x-axis).

(A) Manhattan plot of genome wide association results susceptibility to brucellosis in Damascus goats. Each point represents a SNP. The solid dark blue line represents the threshold line and the second dark red solid line represents the genome-wide significance level for in -log10(P-value) scale in the y-axis and chromosomes are in the x-axis. (B) Manhattan plot of genome wide association results of susceptibility to brucellosis in Damascus goats. Each point represents a SNP. The solid dark blue line represents the threshold line and the second dark red solid line represents the genome-wide significance level for in Fst scale in the y-axis and chromosomes are in the x-axis.
F st results
The top 10 SNPs that accounted for the highest Fst values in the current study (Fig. 3B) were observed on chromosomes 18 (7 Mb), 15 (29 Mb), 2 (2 Mb), 30 (25 Mb), 15 (72 Mb), 26 (34 Mb), 18 (7 Mb), 12 (63 Mb), 3 (36 Mb) and 24 (46 Mb) (Fig. 3) and accounted for Fst values of 0.41, 0.40, 0.39, 0.38, 0.37, 0.36, 0.36, 0.35, 0.34 and 0.33, respectively (Table 2). Information about all suggestive significant SNPs and the Fst estimates explained by each SNP are presented in the supplementary excel file. These identified chromosomal regions harbor candidate genes that encode proteins involved in the immunoglobulin superfamily and autophagy and pathways related to the innate immune system. A considerable overlapping was observed between the highest significantly associated genomic regions from GWAS and Fst approaches implemented in the current study. The common variants from both approaches were observed on Chr 18 (7 Mb) and Chr15 (29 Mb) and these genomic regions harbor important functional candidate genes for the disease.
SNP-based heritability
Genotypic (σ2g), residual (σ2e) and phenotypic (σ2p) variances and corresponding standard errors (SE) were estimated based on genomic information as 0.038 ± 0.039, 0.1 ± 0.04 and 0.14 ± 0.025, respectively. SNP-based h2 for susceptibility to brucellosis infection in goats based on genomic information was estimated as 0.26 ± 0.26.
Genetic variance explained by markers
A genome window was considered to have a significant contribution to the genetic variance of the trait if it accounted for > 1%61. Accordingly, 14 genome windows with a total of 2.32% of genetic variance were identified. These windows were located on chromosomes 14 (17 Mb), 15 (72 Mb), 7 (40 Mb), 21 (46 Mb), 13 (48 Mb), 16 (43 Mb), 18 (15 Mb), 30 (22 Mb), 12 (0 Mb), 6 (84 Mb), 1 (66 Mb), 5 (20 Mb), 23 (4 Mb) and 30 (21 Mb) explaining about 0.47%, 0.43%, 0.2%, 0.12%, 0.12%, 0.12, 0.12%, 0.11%, 0.1%, 0.1%, 0.1%, 0.1%, 0.1% and 0.1% of the genetic variance, respectively (Table 3).
Diversity and composition of vaginal bacteria
The sequencing failed in four vaginal DNA samples therefore fecal samples of the same animals were discarded from the study. The sequencing of 12 samples generated 1,035,511 high-quality sequence reads with a mean of 95,304 ± 11,841 (mean ± SE) reads per sample. The number of ASVs and alpha diversity indices, Chao1, Invsimpson, and Shannon, were greater in vaginal positive (VP) than vaginal negative (VN) group but this difference was not significant (P > 0.05) (Table 4). The PCoA analysis based on Bray Curtis metrics (Fig. 4A) showed that microbial communities of VN and VP were clustered separately. The analysis of the vaginal bacterial community in VN and VP revealed 15 bacterial phyla (Table 5). The bacterial community was dominated by phylum Proteobacteria and Firmicutes, which together constitute about 90% of the bacterial community. Bacterial phyla that represented 1–3.9% were Actinobacteriota and Bacteroidetes. Minor phyla that represented less than 1% were Spirochaetota, Planctomycetota, Deinococcota, Gemmatimonadota, Verrucomicrobiota, Acidobacteriota, Desulfobacterota, Myxococcota and Chloroflexi. Moreover, phylum Deferribacterota and Armatimonadota were detected exclusively in VP group (Table 5). Unclassified bacteria within vaginal bacteria represented more than 17% of the bacterial community (Table 6).

(A) Principal coordinates analysis of vaginal microbial communities of the non-infected (VN) and infected goats (VP) using Bray–Curtis dissimilarity. The red circles are for VN group and blue circles are for VP group. (B) Principal coordinates analysis of fecal microbial communities of the non-infected (FN) and infected goats (FP) using Bray–Curtis dissimilarity. The red circles are for FN group and blue circles are for FP group.
Phylum Proteobacteria represented 47.65% of vaginal bacteria and was dominated by class Gammaproteobacteria and Alphaproteobacteria. On the genus level, phylum Proteobacteria was dominated by Burkholderia-Caballeronia-Paraburkholderia, Alcaligenes, Halomonas, Ralstonia, Delftia, Pseudochrobactrum, Pseudoxanthomonas, Bradyrhizobium and Paracoccus. Only genus Pseudochrobactrum was significantly higher (P < 0.05) in VP group compared to VN group. Additionally, unclassified bacteria in this phylum represented more than 5% (Table 6). The abundance of genus Burkholderia-Caballeronia-Paraburkholderia was declined from 20.4% in VN group to 13.93% in VP group. Genus Alcaligenes abundance was higher in VP group compared to VN.
Several bacterial genera were observed exclusively in a specific group of goats such as genus Neisseria, Azohydromonas, Limnobacter, Pelagibacterium, Qipengyuania, Pseudorhizobium, and Cupriavidus that were found in the VP group (Supplementary File 2). Also, genus Reyranella, Phenylobacterium, Thermomonas, Paracoccus, Noviherbaspirillum, Conchiformibius, Devosia, Sphingomonas, Pseudorhodoferax, Neorhizobium, Shinella, Rubellimicrobium, Pseudoxanthobacter and Ellin6055 were observed in VN group (Supplementary File 2).
Phylum Firmicutes was the second largest phylum, representing 43.2% of the vaginal microbiome, and was classified mainly to class Bacilli and Clostridia (Tables 5 and 6). On the genus level, phylum Firmicutes was dominated by Lysinibacillus, Streptococcus, Salinicoccus, Staphylococcus, UCG-005, and Christensenellaceae R-7 group. The lactobacillus genus represented less than 0.1% and was higher abundance in VN group than VP group. Some bacterial genera that were observed in a specific group within Firmicutes, including Romboutsia, Granulicatella, Gemella, Tumebacillus, Clostridium sensu stricto 6, Coprococcus, Solibacillus, Lachnoclostridium, Blautia, Epulopiscium, and Alkalibacterium that were observed in VP group (Supplementary File 2). Also, genus Aerococcus, Lachnospiraceae NK4B4 group, Weissella, Globicatella, Lachnospiraceae UCG-010, Granulicatella, Alkaliphilus, Fontibacillus, Dorea, Succiniclasticum, Veillonella, Trichococcus, Lachnospiraceae NK3A20 group, Marvinbryantia, Abiotrophia, Turicibacter, Pontibacter, Fibrisoma, Brachybacterium, Blastococcus, and Marmoricola that were found exclusively in VN samples (Supplementary File 2). Phylum Desulfobacterota was significantly (P < 0.05) higher abundance in VP group whereas Chloroflexi was significantly higher abundance in VN group.
Diversity and composition of fecal bacteria
The sequencing of 12 fecal samples resulted in 929,849 reads with a mean of 77,487 ± 8293 (mean ± SE) reads per sample. The number of ASVs as well as alpha diversity indices, Chao1, Inverse Simpson, and Shannon, were significantly (P < 0.05) higher in the cases (fecal positive, FP) than in the control (fecal negative, FN) (Table 7). The PCoA analysis based on Bray Curtis metrics (Fig. 4B) showed that microbial communities of FN and FP were clustered separately.
The fecal bacterial community was affiliated with 11 bacteria phyla; and two archaeal phyla, Halobacterota and Euryarchaeota that were observed only in FP group. The bacterial community was assigned mainly to Bacteroidotes and Firmicutes which represented together about 95%. The phylum Spirochaetota represented 1.7% of the bacterial community. Other minor bacterial phyla were Planctomycetota, Verrucomicrobiota, Desulfobacterota, Fibrobacterota, Proteobacteria, Actinobacteriota, and Cyanobacteria. Phylum Elusimicrobiota was observed only in the FP group (Table 8). Furthermore, unclassified sequence reads represented 0.27% of total sequenced reads.
The phylum Firmicutes dominated the bacterial community (70.41%) and was higher in FP group compared with FN group without significant difference. This phylum was dominated by Christensenellaceae R-7 group and Lachnospiraceae AC2044 group that were significantly declined in abundance (P < 0.05) in FP group compared to FN (Table 7). Genera Phascolarctobacterium and NK4A214 group were significantly higher (P < 0.05) in FP group. Unclassified bacteria within Firmicutes represented 23.6% of the bacterial community (Table 9).
The phylum Bacteroidetes, the second largest phylum in fecal bacterial community (25.79%), was significantly higher abundant in FN group than FP group (Table 8). On the genus level, this phylum was dominated by Rikenellaceae RC9 gut group that was declined in FP group. In addition, genera Bacteroides, Alistipes, and Prevotellaceae UCG-003 were higher abundant in FP group (Table 9). Phylum Spirochaetota, Planctomycetota, and Proteobacteria were higher abundant in FP group compared with FN group (Table 8). Archaeal phylum Halobacterota was classified into genus Methanocorpusculum. Additionally, archaeal phylum Euryarchaeota was further classified to genera Methanobrevibacter and Methanosphaera.
Discussion
The GWAS for brucellosis infection in Egyptian goats identified novel genetic markers, which may contribute to brucellosis susceptibility. It is important to understand the biological background of the candidate genes identified in the current study. The animal's response to the pathogens relies on the induction of multiple cell-mediated immune systems, such as the innate immune (e.g. SLC11A1, TLR1, and TLR4) and cytokine (e.g. IFNGR1, IFNGR2, TNFA) responses62. Hence, polymorphisms in the coding genes or genes of related pathways may contribute to the immune response capacity of the animal (i.e. disease tolerance or susceptibility). The genetic basis for variations in the animal response to the infectious diseases was previously reported in cattle suggesting the feasibility of genetic selection to improve resistance/tolerance to the disease30. In the current study, some of the significantly identified chromosomal regions harbor genes with biological roles in the innate immune response of the animal. For instance, the snp57399-scaffold913-2774950 (rs268288924) on goat chromosome 20 was located in the intronic region of MAP1B (microtubule associated protein 1B) gene, which acts as a positive cofactor in DAPK1 (death associated protein kinase)-mediated autophagic vesicle formation and membrane blebbing63. The snp20448-scaffold202-3614967 (rs268252959) on chromosome 2 mapped to the intronic region of KCNH7 (potassium voltage-gated channel subfamily H member 7) gene. This gene was identified in the plasma membrane as a potassium channel64 to be involved in multiple cellular processes affect the immune system.
Importantly, three potential candidate genes were located within 1 Mb window of the most significant associated SNP on chromosome 15 (snp1723-scaffold1048-1212160, P = 5.01 × 10–7): (1) the arrestin beta 1 (ARRB1) gene, which is involved in Toll-like and IL-1 receptors signaling. This gene was found to be highly expressed in peripheral blood leukocytes and plays a major role in regulating receptor-mediated immune functions65,66. (2) The RELT-TNF receptor (RELT) gene, which activates the NF-kappaB pathway and binds TNF receptor-associated factor 1 (TRAF1). This receptor is capable of stimulating T-cell proliferation in the presence of CD3 signaling, which suggests its regulatory role in immune response67. (3) The autophagy related 16 like (ATG16L2) gene, which is involved in biological pathways related to autophagy. In agreement, several immunity-related genes have been identified for resistance/susceptibility to brucellosis infection in humans, such as IL-1735, IFN-R136, TGF-β137 and IL6 and IL1038. In contrast, four alleles in the TLR5 gene were identified to be correlated with a seroprevalence of brucellosis in the Saanen goats43, as well as the innate immunity genes in cattle68. A haplotype in the PTPRT gene was associated with resistance to Brucella infection in Argentinian goats41. Associations of TNF rs668920841 and INRA111 polymorphisms with caprine brucellosis was also reported69. Additionally, variability at the SLC11A1 locus has been linked to resistance to brucellosis in Algerian goat70. Few genetic variants and candidate genes have been associated with antibody response in feral swine infected with brucellosis42.
Likewise, other significant SNPs were located in intergenic regions of important candidate genes. For instance, ULK1 (Unc-51 Like autophagy activating kinase 1) gene (CHR17, 25 Mb), which is involved in several processes including autophagosome assembly71. The IFI44L and IFI44 (interferon induced proteins) genes at CHR3 (54 Mb) play a critical role in antiviral and antibacterial activity72. They promote macrophage differentiation and facilitate inflammatory cytokine secretions in the immune response to bacterial infection. The snp20448-scaffold202-3614967 (P = 6.86 × 10–5) on chromosome 2 was in close proximity to three candidate genes (Dipeptidyl Peptidase; DPP4, also known as CD26, interferon induced with helicase C domain; IFIH1 and Grancalcin; GCA), which are involved in the immune regulation and response to infections73,74. Similarly, snp21290-scaffold208-1536919 on chromosome 18 at 70 Mb indicated two potential candidate genes: docking protein (DOK4) and transcription factor (MAF), which are involved in the regulation of the immune response induced by T-cells and activation of the expression of IL4 in T helper 2 cells75,76. The window at 75 Mb on chromosome 5 harbored snp32384-scaffold368-1088126 indicating the interleukin 2 receptor subunit beta (IL2RB) gene, which is involved in IL2 and T cell-mediated immune responses62, and the ubiquitin specific peptidase (USP18) gene, which is involved in regulation of the inflammatory response triggered by type I interferon77.
An interesting genomic region on chromosome 26 (34 Mb) included 3 potential candidate genes that are involved in the immune system: (1) Phosphoinositide-3-Kinase Adaptor Protein (PIK3AP1) gene that contributes to B-cell development, activation of PI3K in natural killer cells and TLR signaling pathways, (2) DNA Nucleotidylexotransferase (DNTT) gene, which encodes a protein expressed in malignant pre-B and pre-T lymphocytes during early differentiation78, and (3) B Cell Linker (BLNK) gene that encodes a cytoplasmic linker or adaptor protein and plays a critical role in B-cell development79. Further chromosomal regions were identified on chromosomes 5 (72 MB), 12 (63 Mb) and 24 (46 Mb) comprising contactin (CNTN5), NHL repeat containing (NHLRC3) and ectopic P-Granules 5 autophagy tethering factor (EPG5) genes that encode proteins involved in the immunoglobulin superfamily80, autophagy and pathways related to the innate immune system81. A considerable overlap was observed between GWAS and Fst approaches pointing to chromosomes 18 (7 Mb) and 15 (29 Mb) with important candidate genes for the disease as shown above.
Comparison between our GWAS results and previous reports is difficult due to scarcity of studies addressing the genetic basis of brucellosis in livestock. Additionally, in case of infected animals, the availability of well-defined adequate phenotypes to clearly diagnose the disease are still challenging27. Accordingly, reanalyzing the data with additional phenotypes followed by fine mapping of positional candidate genes may confirm our findings and help to identifying novel candidate genes for brucellosis susceptibility in livestock.
SNP heritability measures the proportion of phenotypic variance explained by all markers without pedigree82. Such type of genomic-based heritability estimates for brucellosis infection in livestock was not reported yet. The pedigree-based h2 estimate was 0.33 for brucellosis infection in Awassi sheep83 was higher than estimated in our study in Damascus goats. This may be because of using different estimation methods, i.e. pedigree-based and genomic-based, respectively. However, the reasonable h2 estimated here means that SNP information captured most of the variance between individuals in context of the studied traits84. Comparably, quite similar estimates (h2 = 0.03–0.28) using either pedigree or genomic information was reported for paratuberculosis in dairy cattle85. This suggests that brucellosis infection may be heritable and could respond well to genetic improvement programs.
Quantitative traits may be affected by a few genes with large or modest effects plus genes with small effects, otherwise, they are affected by many genes with small effects86. In our study, we identified 14 genome windows each explained more than 0.1% of the total genetic variance and all together explained 2.32% of the genetic variance. These regions harbored genes of immunity, which may be suitable candidates for the susceptibility to brucellosis. Interestingly, the genome window located on chromosome 18 (15 Mb) harbored interleukin 17C (IL17-C) and ring finger protein 166 (RNF166) genes that play a crucial role in the innate immunity87 and autophagy87, respectively. Likewise, the genomic window on chromosome 1 (66 Mb) harbored CD86, poly (ADP-ribose) polymerase family (PARP9), and deltex E3 ubiquitin ligase (DTX3L) candidate genes, which are involved in the immune response88,89. Corresponding to Fst results, the region at 72 Mb on chromosome 15 that harbors the CNTN5 gene was identified here to explain 0.43% of genetic variance. This suggests that this region may plays an important role in the disease susceptibility and may be worth to be involved in further investigations. Otherwise, none of the top significant SNPs were reported in the top genome windows that explain > 1% of phenotypic variance, suggesting that the trait is likely controlled by a multiple SNPs with small to medium effects85.
The immune system has a complex bidirectional relationship with the microbiome of the organism. The diversity of the fecal microbial community showed a crucial association with variations in the immune response, which in turn may alter the microbiome90. This is due to the direct effects of the intestinal microbiota on diet fermentation and animal feed efficiency91,92. Genetic polymorphisms were identified to influence the microbiome in humans. Noteworthy, the vaginal and fecal microbiome is significantly associated with animal performance32. For instance, it was reported that dysbiosis in the vaginal microbiome causes inflammation and declines reproductive efficiency34,91,92,93. In goats, no information is available on the vaginal and fecal microbiome and their relations with reproductive diseases such as brucellosis.
Reproductive disorders increase the microbial diversity in the reproductive tract94. This is consistent with our findings in the vaginal and fecal microbial communities and supported by the results of PCoA. In agreement, Lui et al.95 showed that the vaginal microbiome of aborted women showed higher diversity compared to healthy women. Additionally, the vaginal microbiome in this study was dominated by phylum Firmicutes and Proteobacteria, which agrees with previous studies on cattle32 and sheep34. In this study, the vaginal bacteria were dominated by microbes assigned to genera covering Burkholderia, Caballeronia and Paraburkholderia, which were lower abundant in the infected does compared to the non-infected. Consistently, the abundance of these genera was declined in the aborted women patients with recurrent spontaneous abortion (RSA)96. Moreover, corresponding species were observed in the semen microbiome and showed a positive correlation with the healthy status of the genital tract97. Infected animals showed a higher relative abundance of the genus Halomonas, which is a pathogen bacterium95. Genus Alcaligenes was higher abundant in the infected does which agrees with the pathogenic attributes assigned to species of this genus98. Furthermore, Alcaligenes is a drug-resistant genus and susceptible to a specific combination of antibiotics98,99. These findings highlight the importance of investigating the vaginal microbiome in animals with reproductive disorders to characterise the microbial milieu and identify the best suitable antimicrobial treatment. Genera Streptococcus, Salinicoccus and Staphylococcus cause vaginal disorders and abortion, which may explain their higher proportion in the infected does32,100,101. Staphylococcus was previously observed in ewes34 and dominated the vaginal microbiome of goat in estrus synchronization33. Unlike Human vaginal microbiome, Lactobacillus represented a small proportion in the goats’ vaginal microbiome102. Importantly, some of bacterial genera found exclusively in the infected does are pathogens (e.g. genus Neisseria), which may causes sexually transmitted diseases103.
The differences in the diversity and structure of fecal microbiome were significant between infected and non-infected animals. Also, the bacterial community in fecal samples was dominated by phylum Firmicutes and Bacteroidetes, which agrees with previous studies on fecal microbiota in goats and cattle32,104. Most of bacteroidetes’ members are specialized in the degradation of lignocellulose and soluble polysaccharides in the animal gut105, which may be a reason for decreasing the dietary fiber digestion in the infected animals. The dominant genera in the fecal microbiome were Rikenellaceae RC9 gut group and Christensenellaceae R-7 group, which is consistent with previous studies on goat fecal microbiome104 and the rumen microbiome of cattle92. These genera showed higher relative abundances in the non-infected animals. Members of family Christensenellaceae were related to a healthy phenotype in humans106. Christensenellaceae R-7 group abundance was positively correlated with animal health, feed efficiency, animal’s body mass index, rumen digestion and absorption of nutrients, fiber digestion, and protein metabolism; and this genus produces acetic and butyric acids91,92. Moreover, Rikenellaceae RC9 gut group plays a critical role in the digestion of crude fiber92,105. Accordingly, reproductive disorders could impair animal performance and gut fermentation. Specifically, fiberolytic bacteria not only digest dietary fiber but some also represent a barrier against pathogens107.
Study limitations and future perspectives
Generally, identifying the causative variants for a specific disease is challenging108. Despite GWAS highlights signals in the entire genome that help to understand the biology of disease infection, they do not pinpoint the causative variants. There are many limitations of complex traits (e.g. disease susceptibility) analysis with GWAS. The small sample size, low or missing heritability109 and the phenotype definition (e.g. examining different stages of the disease progression) are of these limitations110. Therefore, it is difficult to identify SNPs with larger effects and there is a lot of discrepancies between GWAS results111. Besides, the identified variants do not capture most of the additive genetic variations due to the disease. However, these investigations are important to clarify the biological basis of disease, providing information about the mechanisms underlying the disease process83. In the current study, the small sample size and identification of the infectious status of brucellosis based on serological test only are limiting our results. Future studies with larger sample size are needed to confirm the identified SNPs that arise from a particular biological pathway. Alongside, combining multiple datasets may increase the statistical power and exhibit the overlapped chromosomal regions that was marginally identified in the previous analysis112. This may also encompass interdisciplinary molecular approaches, such as microbiology and PCR-based identification of the causative agent with different isolates to classify and identify the virulent strain of the disease113.
Conclusion
Lack of enough information concerning the genetics of brucellosis in livestock is a real barrier towards animal welfare, diagnosis, vaccination, and management of the disease. The previous studies pointed out polymorphisms in immunity-related genes as putative candidate genes for disease susceptibility in humans and livestock. Accordingly, the identified variants and candidate genes from our GWAS analysis may contribute to the phenotypic variations between cases and controls of Brucella infection. This included chromosome 15 (29 Mb; ARRB1, RELT and ATG16L2), 20 (9 Mb; MAP1B), 2 (102 Mb; KCNH7), 17 (25 Mb, ULK1), 3 (54 Mb; IFI44L and IFI44), 18 (7 Mb; DOK4 and MAF), 5 (75 Mb; IL2RB) and 26 (34 Mb; PIK3AP1, DNTT, BLNK) as the most important genomic regions potentially contributing to the incidence of brucellosis in the examined goat population. Some of the identified QTLs were overlapped between Fst and GWAS approaches (CHR18: 7 Mb and CHR15: 29 Mb) and all of them are novel. Furthermore, our results suggested that brucellosis infection is heritable (h2 = 0.26) and may respond well to genetic improvement programs. The GWAS analysis may support the development of markers to be used as useful adjunct in controlling the disease. In addition, new perspectives into the interaction between the infection and the diversity and composition of gut and vaginal were obtained.
Data availability
All results are presented in supplementary files. The raw sequence reads of vaginal and fecal microbiome were deposited to the SRA at https://www.ncbi.nlm.nih.gov/sra/PRJNA910086.
References
Cárdenas, L., Peña, M., Melo, O. & Casal, J. Risk factors for new bovine brucellosis infections in Colombian herds. BMC Vet. Res. 15, 81 (2019).
Ko, J. & Splitter, G. A. Molecular host-pathogen interaction in brucellosis: Current understanding and future approaches to vaccine development for mice and humans. Clin. Microbiol. Rev. 16, 65–78 (2003).
Wareth, G., Melzer, F., Tomaso, H., Roesler, U. & Neubauer, H. Detection of Brucella abortus DNA in aborted goats and sheep in Egypt by real-time PCR Veterinary Research. BMC Res. Notes https://doi.org/10.1186/s13104-015-1173-1 (2015).
WHO. The Control of Neglected Zoonotic Diseases—A Route to Poverty Alleviation (WHO, 2006).
Franc, K. A., Krecek, R. C., Häsler, B. N. & Arenas-Gamboa, A. M. Brucellosis remains a neglected disease in the developing world: A call for interdisciplinary action. BMC Public Health 18, 125 (2018).
Corbel, M. Brucellosis in Humans and Animals (World Health Organization, 2006).
Rossetti, C. A., Arenas-Gamboa, A. M. & Maurizio, E. Caprine brucellosis: A historically neglected disease with significant impact on public health. PLoS Negl. Trop. Dis. 11, e0005692–e0005692 (2017).
Bamaiyi, P., Khairani-Bejo, S. & Zainal, A. The economic impact attributable to brucellosis among goat farms in Peninsula Malaysia and cost benefit analysis. Res. Opin. Anim. Vet. Sci. 5, 57–64 (2014).
Deka, R. P., Magnusson, U., Grace, D. & Lindahl, J. Bovine brucellosis: Prevalence, risk factors, economic cost and control options with particular reference to India-a review. Infect. Ecol. Epidemiol. https://doi.org/10.1080/20008686.2018.1556548 (2018).
Lokamar, P. N., Kutwah, M. A., Atieli, H., Gumo, S. & Ouma, C. Socio-economic impacts of brucellosis on livestock production and reproduction performance in Koibatek and Marigat regions, Baringo County, Kenya. BMC Vet. Res. 16, 61 (2020).
Johansen, T. B., Scheffer, L., Jensen, V. K., Bohlin, J. & Feruglio, S. L. Whole-genome sequencing and antimicrobial resistance in Brucella melitensis from a Norwegian perspective. Sci. Rep. 8, 8538 (2018).
Khurana, S. K. et al. Bovine brucellosis—A comprehensive review. Vet. Q. 41, 61–88 (2021).
Ducrotoy, M. J. et al. Brucellosis as an emerging threat in developing economies: Lessons from Nigeria. PLoS Negl. Trop. Dis. 8, e3008 (2014).
Terefe, Y., Girma, S., Mekonnen, N. & Asrade, B. Brucellosis and associated risk factors in dairy cattle of eastern Ethiopia. Trop. Anim. Health Prod. 49, 599–606 (2017).
Gibbs, J. & Bercovich, Z. Diseases of dairy animals | infectious diseases: Brucellosis. In Encyclopedia of Dairy Sciences 2nd edn (ed. Fuquay, J. W.) 153–159 (Academic Press, 2011). https://doi.org/10.1016/B978-0-12-374407-4.00131-X.
Akhtar, R. et al. Genotyping of Brucella strains isolated from humans and cattle of different geographical regions of Pakistan using MLVA-15. Vet. Med. Sci. 7, 1688–1695 (2021).
FAO. Brucella melitensis en Eurasia and the Middle East (2010).
El-Moselhy, E. et al. Human brucellosis: Methods of diagnosis and risk factors among Egyptian patients admitted at Assiut fever hospital. J. Recent Adv. Med. 1, 1–13 (2020).
Eltholth, M. M., Hegazy, Y. M., El-Tras, W. F., Bruce, M. & Rushton, J. Temporal analysis and costs of ruminant brucellosis control programme in Egypt between 1999 and 2011. Transbound. Emerg. Dis. 64, 1191–1199 (2017).
Refai, M. Review: Incidence and control of brucellosis in the Near East region. Vet. Microbiol. 90, 81–110 (2002).
Samaha, H., Al-Rowaily, M., Khoudair, R. M. & Ashour, H. M. Multicenter study of brucellosis in Egypt. Emerg. Infect. Dis. 14, 1916–1918 (2008).
Hegazy, Y. M., Moawad, A., Osman, S., Ridler, A. & Guitian, J. Ruminant brucellosis in the Kafr El Sheikh governorate of the Nile Delta, Egypt: Prevalence of a neglected zoonosis. PLoS Negl. Trop. Dis. 5, e944 (2011).
Musallam, I. I., Abo-Shehada, M. N., Hegazy, Y. M., Holt, H. R. & Guitian, F. J. Systematic review of brucellosis in the Middle East: Disease frequency in ruminants and humans and risk factors for human infection. Epidemiol. Infect. 144, 671–685. https://doi.org/10.1017/S0950268815002575 (2016).
Li, T. et al. Brucella melitensis M5–90Δbp26 as a potential live vaccine that allows for the distinction between natural infection and immunization. Can. J. Microbiol. 63, 719–729 (2017).
Mandal, S. S. et al. Novel solutions for vaccines and diagnostics to combat Brucellosis. ACS Cent. Sci. 3, 224–231 (2017).
Ahmed, M., Mostafa, A. E. & Abou Gazia, K. Conventional, serological and molecular characterization of Brucella species isolated from different governorates in Egypt. J. Curr. Vet. Res. 3, 107–117 (2021).
Schaid, D. J., Chen, W. & Larson, N. B. From genome-wide associations to candidate causal variants by statistical fine-mapping. Nat. Rev. Genet. 19, 491–504 (2018).
Jiang, J. et al. Functional annotation and Bayesian fine-mapping reveals candidate genes for important agronomic traits in Holstein bulls. Commun. Biol. 2, 1–12 (2019).
Freebern, E. et al. GWAS and fine-mapping of livability and six disease traits in Holstein cattle. BMC Genom. 21, 41 (2020).
Berry, D. P., Bermingham, M. L., Good, M. & More, S. J. Genetics of animal health and disease in cattle. Ir. Vet. J. https://doi.org/10.1186/2046-0481-64-5 (2011).
Sallam, A. M., Zare, Y., Shook, G., Collins, M. & Kirkpatrick, B. W. A positional candidate gene association analysis of susceptibility to paratuberculosis on bovine chromosome 7. Infect. Genet. Evol. 65, 163–169 (2018).
Deng, F. et al. The vaginal and fecal microbiomes are related to pregnancy status in beef heifers. J. Anim. Sci. Biotechnol. 10, 92 (2019).
Oliveira, J. K. et al. Changes in the vaginal flora of goats following a short-term protocol of oestrus induction and synchronisation with intravaginal sponges as well as their antimicrobial sensitivity. Small Rumin. Res. 113, 162–166 (2013).
Greenwood, E. C., Torok, V. A. & van Wettere, W. H. E. J. Characterizing the vaginal microbiota of high and low producing Poll Merino and White Suffolk ewes. Transl. Anim. Sci. https://doi.org/10.1093/tas/txac133 (2022).
Rasouli, M., Asaei, S., Kalani, M., Kiany, S. & Moravej, A. Interleukin-17A genetic variants can confer resistance to brucellosis in Iranian population. Cytokine 61, 297–303 (2013).
Naseri, Z., Bahmani, N., Alikhani, M. Y., Hashemi, S. H. & Roshanaei, G. Polymorphisms in promoter region of the interferon—Gamma Receptor1 gene and its relation with susceptibility to brucellosis. Iran. J. Pathol. 14, 206–211 (2019).
Fu, J. et al. Association of IL-6, IL-10 and TGF-β1 gene polymorphisms with brucellosis: A systematic review with meta-analysis. Microb. Pathog. 135, 103640 (2019).
Jin, X., Wu, Y., Yin, S., Chen, X. & Zhang, Y. Association between the IL-10 and IL-6 polymorphisms and brucellosis susceptibility: A meta-analysis. BMC Med. Genet. 21, 63 (2020).
Martínez, R. et al. Effect of polymorphisms in the Slc11a1 coding region on resistance to brucellosis by macrophages in vitro and after challenge in two Bos breeds (Blanco Orejinegro and Zebu). Genet. Mol. Biol. 33, 463–470 (2010).
Iacoboni, P. A. et al. Polymorphisms at the 3′ untranslated region of SLC11A1 gene are associated with protection to Brucella infection in goats. Vet. Immunol. Immunopathol. 160, 230–234 (2014).
Rossi, U. A. et al. A haplotype at intron 8 of PTPRT gene is associated with resistance to Brucella infection in Argentinian creole goats. Vet. Microbiol. 207, 133–137 (2017).
Pierce, C. F. et al. Loci associated with antibody response in feral swine (Sus scrofa) infected with Brucella suis. Front. Vet. Sci. 7, 957 (2020).
Wang, X. et al. Nucleotide variants in TheTLR5 gene and promoter methylation with A susceptibility to brucellosis in Chinese goats. Folia Biol. (Kraków) 70, 55–66 (2022).
Adams, L. G. & Schutta, C. J. Natural resistance against brucellosis: A review. Open Vet. Sci. J. 4, 61–71 (2010).
Rodrigues, N. F. et al. Qualitative analysis of the vaginal microbiota of healthy cattle and cattle with genital-tract disease. Genet. Mol. Res. 14, 6518–6528 (2015).
Díaz, R., Casanova, A., Ariza, J. & Moriyón, I. The rose bengal test in human brucellosis: A neglected test for the diagnosis of a neglected disease. PLoS Negl. Trop. Dis. 5, e950 (2011).
Park, S.-H. et al. Application of the microagglutination test for serologic diagnosis of human brucellosis. Osong Public Health Res. Perspect. 3, 19–23 (2012).
Chang, C. C. et al. Second-generation PLINK: Rising to the challenge of larger and richer datasets. Gigascience https://doi.org/10.1186/s13742-015-0047-8 (2015).
Zheng, X. et al. A high-performance computing toolset for relatedness and principal component analysis of SNP data. Bioinformatics 28, 3326–3328 (2012).
Burton, P. R. et al. Genome-wide association study of 14,000 cases of seven common diseases and 3000 shared controls. Nature 447, 661–678 (2007).
Duggal, P., Gillanders, E. M., Holmes, T. N. & Bailey-Wilson, J. E. Establishing an adjusted p-value threshold to control the family-wide type 1 error in genome wide association studies. BMC Genom. 9, 516 (2008).
Turner, S. qqman: An R package for visualizing GWAS results using Q-Q and manhattan plots. J. Open Sour. Softw. 3, 731 (2018).
Yang, J. et al. Common SNPs explain a large proportion of the heritability for human height. Nat. Genet. 42, 565–569 (2010).
Legarra, A., Christensen, O. F., Aguilar, I. & Misztal, I. Single step, a general approach for genomic selection. Livest. Sci. 166, 54–65 (2014).
Aguilar, I. et al. Hot topic: A unified approach to utilize phenotypic, full pedigree, and genomic information for genetic evaluation of Holstein final score1. J. Dairy Sci. 93, 743–752 (2010).
Zhang, Z. et al. Best linear unbiased prediction of genomic breeding values using a trait-specific marker-derived relationship matrix. PLoS One 5, e12648 (2010).
Wang, H., Huang, J., Xu, X. & Xiao, Y. Damage attack on complex networks. Phys. A Stat. Mech. Appl. 408, 134–148 (2014).
Kinsella, R. J. et al. Ensembl BioMarts: A hub for data retrieval across taxonomic space. Database 2011, 1–9 (2011).
Safran, M. et al. The GeneCards suite. In Practical Guide to Life Science Databases (eds Abugessaisa, I. & Kasukawa, T.) 27–56 (Springer Nature Singapore, 2021). https://doi.org/10.1007/978-981-16-5812-9_2.
Callahan, B. J. et al. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583 (2016).
Peters, S. O. et al. Bayesian genome-wide association analysis of growth and yearling ultrasound measures of carcass traits in brangus heifers. J. Anim. Sci. 90, 3398–3409 (2012).
Aristizábal, B. & González, Á. Innate immunity system. In Autoimmunity: From Bench to Bedside (eds Juan-Manuel, A. et al.) 237–839 (El Rosario University Press, 2013).
Harrison, B. et al. DAPK-1 binding to a linear peptide motif in MAP1B stimulates autophagy and membrane blebbing*. J. Biol. Chem. 283, 9999–10014 (2008).
Oshiro, C., Thorn, C. F., Roden, D. M., Klein, T. E. & Altman, R. B. KCNH2 pharmacogenomics summary. Pharmacogenet. Genom. 20, 775–777 (2010).
Wang, Y. et al. Association of β-arrestin and TRAF6 negatively regulates Toll-like receptor–interleukin 1 receptor signaling. Nat. Immunol. 7, 139–147 (2006).
Li, H. et al. Arrestin 2 negatively regulates toll-like receptor 4 (TLR4)-triggered inflammatory signaling via targeting p38 MAPK and interleukin 10 *. J. Biol. Chem. 289, 23075–23085 (2014).
Yao, W. et al. RELT promotes the growth of esophageal squamous cell carcinoma by activating the NF-κB pathway. Cell Cycle 20, 1231–1241 (2021).
Prakash, O. et al. Polymorphism of cytokine and innate immunity genes associated with bovine brucellosis in cattle. Mol. Biol. Rep. 41, 2815–2825 (2014).
Hasenauer, F. C. et al. Association of TNF rs668920841 and INRA111 polymorphisms with caprine brucellosis: A case-control study of candidate genes involved in innate immunity. Genomics 112, 3925–3932 (2020).
Saahraoui, H. et al. Genetic variability in the A microsatellite at SLC11A1 gene and possible implications with innate resistance against brucellosis in Algerian native goats. Biodiversitas 21, 5630–5636 (2020).
Zou, L. et al. Autophagy and beyond: Unraveling the complexity of UNC-51-like kinase 1 (ULK1) from biological functions to therapeutic implications. Acta Pharm. Sin. B 12, 3743–3782 (2022).
Jiang, H., Tsang, L., Wang, H. & Liu, C. IFI44L as a forward regulator enhancing host antituberculosis responses. J. Immunol. Res. 2021, 5599408 (2021).
Shao, S., Xu, Q., Yu, X., Pan, R. & Chen, Y. Dipeptidyl peptidase 4 inhibitors and their potential immune modulatory functions. Pharmacol. Ther. 209, 107503 (2020).
Kim, T. W., Hong, S., Talukder, A. H., Pascual, V. & Liu, Y.-J. Grancalcin (GCA) modulates Toll-like receptor 9 (TLR9) mediated signaling through its direct interaction with TLR9. Eur. J. Immunol. 46, 712–724 (2016).
Gérard, A. et al. Dok-4 is a novel negative regulator of T cell activation. J. Immunol. 182, 7681 (2009).
Gabryšová, L. et al. c-Maf controls immune responses by regulating disease-specific gene networks and repressing IL-2 in CD4+ T cells. Nat. Immunol. 19, 497–507 (2018).
Hou, J. et al. USP18 positively regulates innate antiviral immunity by promoting K63-linked polyubiquitination of MAVS. Nat. Commun. 12, 2970 (2021).
Rothenberg, E. V. Transcriptional control of early T and B cell developmental choices. Annu. Rev. Immunol. 32, 283–321 (2014).
Minegishi, Y. et al. An essential role for BLNK in human B cell development. Science 286, 1954–1957 (1999).
Kamei, Y. et al. Human NB-2 of the contactin subgroup molecules: Chromosomal localization of the gene (CNTN5) and distinct expression pattern from other subgroup members. Genomics 69, 113–119 (2000).
Piano Mortari, E. et al. The Vici syndrome protein EPG5 regulates intracellular nucleic acid trafficking linking autophagy to innate and adaptive immunity. Autophagy 14, 22–37 (2018).
Zhu, H. & Zhou, X. Statistical methods for SNP heritability estimation and partition: A review. Comput. Struct. Biotechnol. J. 18, 1557–1568 (2020).
Sallam, A. M. et al. An across-breed genome wide association analysis of susceptibility to paratuberculosis in dairy cattle. J. Dairy Res. https://doi.org/10.1017/S0022029916000807 (2017).
Zare, Y., Shook, G. E., Collins, M. T. & Kirkpatrick, B. W. Genome-wide association analysis and genomic prediction of Mycobacterium avium subspecies paratuberculosis infection in US Jersey cattle. PLoS One 9, e88380 (2014).
Zhang, F. et al. Genetic architecture of quantitative traits in beef cattle revealed by genome wide association studies of imputed whole genome sequence variants: I: Feed efficiency and component traits. BMC Genom. 21, 1–22 (2020).
Zenobia, C. & Hajishengallis, G. Basic biology and role of interleukin-17 in immunity and inflammation. Periodontology 69, 142–159 (2015).
Chen, H.-W. et al. Ring finger protein 166 potentiates RNA virus-induced interferon-β production via enhancing the ubiquitination of TRAF3 and TRAF6. Sci. Rep. 5, 14770 (2015).
Barclay, A. N. Immunoglobin superfamily. In Brenner’s Encyclopedia of Genetics 2nd edn (eds Maloy, S. & Hughes, K.) 29–31 (Academic Press, 2013). https://doi.org/10.1016/B978-0-12-374984-0.00772-5.
Zhu, H., Tang, Y.-D., Zhan, G., Su, C. & Zheng, C. The critical role of PARPs in regulating innate immune responses. Front. Immunol. https://doi.org/10.3389/fimmu.2021.712556 (2021).
Awany, D. et al. Host and microbiome genome-wide association studies: Current state and challenges. Front. Genet. https://doi.org/10.3389/fgene.2018.00637 (2019).
Bach, A. et al. Changes in the rumen and colon microbiota and effects of live yeast dietary supplementation during the transition from the dry period to lactation of dairy cows. J. Dairy Sci. 102, 6180–6198 (2019).
Huang, C. et al. Microbiome and metabolomics reveal the effects of different feeding systems on the growth and ruminal development of yaks. Front. Microbiol. https://doi.org/10.3389/fmicb.2021.682989 (2021).
Gilbert, R. O., Shin, S. T., Guard, C. L., Erb, H. N. & Frajblat, M. Prevalence of endometritis and its effects on reproductive performance of dairy cows. Theriogenology 64, 1879–1888 (2005).
Oakley, B. B., Fiedler, T. L., Marrazzo, J. M. & Fredricks, D. N. Diversity of human vaginal bacterial communities and associations with clinically defined bacterial vaginosis. Appl. Environ. Microbiol. 74, 4898–4909 (2008).
Liu, X. et al. Association between vaginal microbiota and risk of early pregnancy miscarriage. Comp. Immunol. Microbiol. Infect. Dis. 77, 101669 (2021).
Zhao, F. et al. Characterization of vaginal microbiota in women with recurrent spontaneous abortion that can be modified by drug treatment. Front. Cell. Infect. Microbiol. https://doi.org/10.3389/fcimb.2021.680643 (2021).
Yao, Y. et al. Semen microbiota in normal and leukocytospermic males. Asian J. Androl. 24, 398 (2022).
Saiman, L. et al. Identification and antimicrobial susceptibility of Alcaligenes xylosoxidans isolated from patients with cystic fibrosis. J. Clin. Microbiol. 39, 3942–3945 (2001).
Huang, C. Extensively drug-resistant Alcaligenes faecalis infection. BMC Infect. Dis. 20, 833 (2020).
Maniatis, A. N. et al. Streptococcus agalactiae: A vaginal pathogen?. J. Med. Microbiol. 44, 199–202 (1996).
Sun, D. et al. The association between vaginal microbiota disorders and early missed abortion: A prospective study. Acta Obstet. Gynecol. Scand. 101, 960–971 (2022).
Parolin, C. et al. Insights into vaginal bacterial communities and metabolic profiles of chlamydia trachomatis infection: Positioning between eubiosis and dysbiosis. Front. Microbiol. https://doi.org/10.3389/fmicb.2018.00600 (2018).
Hooda, Y., Shin, H. E., Bateman, T. J. & Moraes, T. F. Neisserial surface lipoproteins: Structure, function and biogenesis. Pathog. Dis. 75, ftx010 (2017).
Mahayri, T. M. et al. Host species affects bacterial evenness, but not diversity: Comparison of fecal bacteria of cows and goats offered the same diet. Animals 12, 2011 (2022).
Rabee, A. E., Forster, R., Elekwachi, C., Sabra, E. & Lamara, M. Comparative analysis of the metabolically active microbial communities in the rumen of dromedary camels under different feeding systems using total rRNA sequencing. PeerJ 8, e10184 (2020).
Waters, J. L. & Ley, R. E. The human gut bacteria Christensenellaceae are widespread, heritable, and associated with health. BMC Biol. 17, 83 (2019).
Rabee, A. E., Younan, B. R., Kewan, K. Z., Sabra, E. A. & Lamara, M. Modulation of rumen bacterial community and feed utilization in camel and sheep using combined supplementation of live yeast and microalgae. Sci. Rep. 12, 12990 (2022).
Berry, D., Bermingham, M., Good, M. & More, S. Genetics of animal health and disease in cattle. Ir. Vet. J. https://doi.org/10.1186/2046-0481-64-5 (2011).
Witte, J. S. Genome-wide association studies and beyond. Annu. Rev. Public Health 31, 9–20 (2010).
Minozzi, G. et al. Meta-analysis of two genome-wide association studies of bovine paratuberculosis. PLoS One 7, e32578 (2012).
Kirkpatrick, B. W. & Shook, G. E. Genetic susceptibility to paratuberculosis. Vet. Clin. Food Anim. 27, 559–571 (2011).
van den Berg, I. et al. Meta-analysis for milk fat and protein percentage using imputed sequence variant genotypes in 94,321 cattle from eight cattle breeds. Genet. Sel. Evol. 52, 37 (2020).
Vouga, M. & Greub, G. Emerging bacterial pathogens: The past and beyond. Clin. Microbiol. Infect. 22, 12–21 (2016).
Acknowledgements
This work was supported by grants from the Science, Technology & Innovation Funding Authority (STDF), the Egyptian Ministry for Scientific Research, within the activities of the ongoing research project with reference number “43213”.
Funding
Open access funding provided by The Science, Technology & Innovation Funding Authority (STDF) in cooperation with The Egyptian Knowledge Bank (EKB). The funding was supported by Science and Technology Development Funding Authority (STDF), the Egyptian Ministry for Scientific Research, within the activities of the research project with reference number “4321”.
Author information
Authors and Affiliations
Contributions
A.M.S. analyzed the GWAS data, interpreted the results and wrote the manuscript. I.A. preprepared DNA samples for genotyping and revised the manuscript. H.R. involved in genotyping, data analysis and revised the manuscript. K.W. has critically edited and revised the manuscript and A.E.R. analyzed the microbiome data, interpreting the results and wrote the manuscript. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sallam, A.M., Abou-souliman, I., Reyer, H. et al. New insights into the genetic predisposition of brucellosis and its effect on the gut and vaginal microbiota in goats. Sci Rep 13, 20086 (2023). https://doi.org/10.1038/s41598-023-46997-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-46997-x
This article is cited by
-
Rumen fermentation and microbiota in Shami goats fed on condensed tannins or herbal mixture
BMC Veterinary Research (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
