
Abstract
Anthropogenic atmospheric pollution and immune response regularly expose bacteria to toxic nitrogen oxides such as NO• and NO2. These reactive molecules can damage a wide variety of biomolecules such as DNA, proteins and lipids. Several components of the bacterial envelope are susceptible to be damaged by reactive nitrogen species. Furthermore, the hydrophobic core of the membranes favors the reactivity of nitrogen oxides with other molecules, making membranes an important factor in the chemistry of nitrosative stress. Since bacteria are often exposed to endogenous or exogenous nitrogen oxides, they have acquired protection mechanisms against the deleterious effects of these molecules. By exposing bacteria to gaseous NO2, this work aims to analyze the physiological effects of NO2 on the cell envelope of the airborne bacterium Pseudomonas fluorescens MFAF76a and its potential adaptive responses. Electron microscopy showed that exposure to NO2 leads to morphological alterations of the cell envelope. Furthermore, the proteomic profiling data revealed that these cell envelope alterations might be partly explained by modifications of the synthesis pathways of multiple cell envelope components, such as peptidoglycan, lipid A, and phospholipids. Together these results provide important insights into the potential adaptive responses to NO2 exposure in P. fluorescens MFAF76a needing further investigations.
Similar content being viewed by others

Introduction
Reactive nitrogen species (RNS) form a group of highly reactive compounds that include nitric oxide (NO•), nitrogen dioxide (NO2) and peroxynitrite (ONOO−). NO• and NO2 are major pollutants resulting from fuel combustion processes used in the industry and transport. These molecules are responsible for environmental problems such as acid rain, photochemical smog and ozone depletion1. RNS can react with a wide range of biological molecules, such as DNA2, proteins3 and lipids4,5. Indeed, NO• can induce nitrosative deamination of DNA bases and sugar oxidative modifications of DNA2. Meanwhile, peroxynitrite can act as a strong oxidant toward thiols and transition metal centers, and can induce tyrosine nitration3. Furthermore, NO2 has the ability to induce lipid peroxidation5. This ability to damage biomolecules makes RNS toxic for biological organisms at high concentrations. RNS concentration in the atmosphere is highly dependent on the local polluting human activities as well as the climatic conditions6,7,8. For human health, the World Health Organization fixes the annual guideline value at 0.1 ppm, and NO2 is considered to have reversible effects on human health after 1 h of exposition to 5 ppm, and irreversible effects after 1 h at 45 ppm9,10.
In the environment, microorganisms can be exposed to RNS from various sources, such as the atmosphere or the immune response, as well as collateral products of their own metabolism11,12. Some bacterial species possess denitrification pathways allowing them to use these species as terminal electron acceptors for their energetic metabolism12,13,14. The mechanisms developed by bacteria to counteract the effects of RNS are tightly linked to their virulence. Indeed, during the primary immune response, macrophages produce a burst of RNS and reactive oxygen species (ROS) to eliminate pathogens11. Thus, resistance to RNS is often a crucial part of the infectious process15,16.The first bacterial component to encounter exogenous RNS is the bacterial envelope. In Gram negative bacteria, this envelope is constituted by two lipid bilayers separated by the periplasmic space containing the peptidoglycan layer. The stability and functions of these membranes are the result of complex interactions between lipids and proteins. Indeed, membrane proteins functionality is often dependent on the steric hindrance caused by nearby lipids17,18. Membranes play an important role in the context of nitrosative stress4. NO• and NO2, the most common RNS found in the environment, is more soluble in the hydrophobic core of the membranes than in water4. Their great concentration in the membrane enhances their reactivity with other RNS and reactive oxygen species (ROS) such as O2●−, which reacts with NO• to form the extremely reactive ONOO−19. Furthermore, NO2 can react with fatty acids possessing at least two unsaturations in several ways such as peroxidation, nitration and nitrosylation4.
Pseudomonas fluorescens is considered a ubiquitous bacterium with a very adaptable metabolism. The P. fluorescens MFAF76a strain was isolated from air and possesses several virulence factors, such as the secretion of lipases and proteases, the ability to form biofilms at 37 °C and a high lytic activity on the human pulmonary cell line A54920. These characteristics make the strain MFAF76a a good model to study the effects of atmospheric NO2 pollution on bacteria. The objectives of this study were to assess the physiological impact of gaseous NO2 on the bacterial cell envelope and to characterize the potential molecular mechanisms underlying the adaptive responses to NO2 exposure in P. fluorescens MFAF76a.
Materials and methods
Bacterial strain
The bacterial strain used in this study is the Gram negative P. fluorescens MFAF76a, an airborne strain isolated from dust clouds of the Rouen harbor installations (Normandy, France) generated during crop ship loading20.
Exposition to NO2
P. fluorescens strain MFAF76a was precultured at 28 °C for 24 h in Lysogeny Broth (LB) (yeast extract 5 g/L, peptone 10 g/L, NaCl 5 g/L). Precultures were used to inoculate strain MFAF76a at a final optical density at 580 nm (OD580) of 0.08 in Davis Medium Broth (DMB) supplemented with 2.16 g/L of glucose21. Culture was incubated at 28 °C during 16 h with agitation at 180 rpm. Bacteria were then centrifuged at 13,000 g for 10 min at 4 °C and resuspended in DMB at an OD580 nm value of 4. Then, 125 µL of bacteria were deposited on nitrocellulose filters (Pore size 0.2 µm, diameter 47 mm, Sartorius Stedim Biotech GmbH, Germany) themselves deposited on Petri dishes containing DMB and 15 g/L agar. Bacteria were incubated 4 h at 28 °C and the filters were deposited on 15 g/mL agar plates, and placed into the exposition system described by Kondakova and coworkers21. Bacteria were exposed to 45 ppm of gaseous NO2 at a N2/O2 ratio of 8/2 (v/v) or synthetic air with the same N2/O2 ratio for 2 h with a constant gas stream of 2 L/min. Exposition to NO2 and air were limited to 2 h to prevent excessive drying of the cells, as well as for time constraints. After exposure, bacteria were resuspended in sterile physiological water (NaCl 9 g/L). The collected exposed (45 ppm NO2) and control (synthetic air) bacterial suspensions were then used for all subsequent experiments.
Electronic microscopy
The sample preparation method for electronic microscopy was adapted from Bergeau et al.22. After exposition to NO2 or synthetic air, 1 mL of bacterial suspension was centrifuged 5 min at 8,000 g at room temperature (RT). Supernatant was discarded and bacteria were resuspended into 100 µL of PBS. 50 µL were deposited onto electron microscopy grids for 5 min. The suspension was discarded, and the cells were fixated for 20 min with 50 µL of fixation solution (Paraformaldehyde 2%, Glutaraldehyde 0.2%, PBS). Bacteria were then washed three times by PBS for 5 min and washed three times with deionized water. Fixated bacteria were then observed by scanning electronic microscopy (JEOL JCM-6000, Tokyo, Japan). Bacteria with apparent envelope alterations were manually counted using ImageJ software version 1.52a (5 images were taken per experiment, N = 3), and their number was divided by the total number of cells23. Only cells that were fully visible on the pictures were taken into account.
Flow cytometry assay
The exposed and control bacterial suspensions after exposure were stained for 15 min with propidium iodine 30 nM (PI) and SYTO9™ 5.01 nM using the Live/Dead BacLight kit (L-7012, Thermofisher, United States) according to the manufacturer instructions. Bacteria were observed using flow cytometry (CytoFLEX S, Beckman Coulter, United States) and the Cytexpert v1.2 software. PI and SYTO9™ were excited at a wavelength of 488 nm and their fluorescence emissions were detected at 690 ± 50 nm and 525 ± 40 nm, respectively. A total of 10,000 events were collected for each measurement at a low flow (10 µL/min). Positive control for membrane integrity was performed on bacteria by treating bacteria with 50% ethanol for 10 min prior to staining. The software was calibrated using the positive integrity control.
Whole proteome analysis
The bacterial suspension after exposition was centrifuged at 13,000 g at 4 °C for 20 min. Pellets were then resuspended in 20 mM Tris–HCl buffer (pH 7.4) and sonicated. Bacterial lysate was centrifuged at 10,000 g at 4 °C for 10 min, and their protein concentration was determined by Bradford test (Biorad™, United States). Total proteins were identified as described previously24. Briefly, proteins were loaded onto an SDS-PAGE stacking gel (7%). After short electrophoresis migration, proteins were reduced, alkylated and digested with trypsin. After extraction, peptides were injected on a LTQ-Orbitrap Elite mass spectrometer coupled to an Easy nLC II system (Thermo Scientific, United States). The mobile phase was composed of 0.1% formic acid (FA) in H2O (buffer A) and 0.1% FA in acetonitrile (buffer B). Samples were injected onto an enrichment column (C18 Acclaim PepMap100, Thermo Scientific, United States) and eluted using a three-step linear gradient (from 2 to 40% B over 75 min, from 40 to 80% B in 4 min, and 11 min at 80% B) at a flow rate of 300 nL/min. The samples were analyzed using CID (collision induced dissociation) method. All measurements in the Orbitrap analyzer were performed with on-the-fly internal recalibration (lock mass) at m/z 445.12002 (polydimethylcyclosiloxane). After MS analysis, raw data were imported in Progenesis LC–MS software (Nonlinear Dynamics, Netherlands). For comparison, one sample was set as a reference and the retention times of all other samples within the experiment were aligned. After the statistical analysis was performed, peptide features presenting a p-value and a q-value less than 0.05, and a power greater than 0.8 were retained. MS/MS spectra from selected peptides were exported for peptide identification with Mascot (Matrix Science, United Kingdom) against the database restricted to P. fluorescens A506 (http://www.pseudomonas.com).
Membrane fluidity assay
The exposed (NO2 45 ppm) and control (synthetic air) bacterial suspensions were washed 3 times with MgSO4 10 mM by centrifugation at 13,000 g for 5 min at RT and resuspension of the pellets in a solution of 10 mM of MgSO4. Bacteria were then resuspended in MgSO4 10 mM and adjusted to an OD580 value of 0.1. Next, bacterial membranes were labelled using 1,6-diphenyl-1,3,5-hexatriene (DPH) fluorescent probe at a final concentration of 4 µM of DPH and incubated in darkness at 28 °C for 30 min. Fluorescence anisotropy was measured with a Spark 20 M multimode microplate reader (Tecan Group Ltd., Männedorf, Switzerland Inc., Research Triangle Park, N.C.). Steady state anisotropy was calculated using Eq. (1):
where I represents fluorescence intensity, and “v” and “h” respectively represent vertical and horizontal settings of the excitation and emission polarizers. The instrumental correction factor G was calculated separately for each experiment by Eq. (2):
Quantitative reverse-transcription real time polymerase chain reaction (RT-qPCR)
Total RNAs were isolated either from pellets of bacteria exposed (45 ppm NO2) or control (synthetic air) using the hot acid-phenol method25. Residual genomic DNA was eliminated using Turbo DNA-free kit (Invitrogen, United States). RNAs were non-specifically converted to single stranded cDNAs using the High-Capacity cDNA Reverse Transcription Kit following manufacturer’s protocol (Applied Biosystems, United States). RT-qPCR experiments were performed as previously described26 using the primers listed in Table 1. The relative quantification of the mRNAs was obtained by the comparative CT (2–ΔΔCT) method as described in27. Measurement of recA gene expression was used as an internal standard. recA has stable expression within the samples to be compared, regardless of physiological or experimental conditions28.
Statistical analyses
All experiments were repeated at least three times. Mann–Whitney test was used to determine the significance between mean values of the cell sizes obtained by flow cytometry. Two tailed unpaired t-test was used to determine the significances of differences between mean values for the live/dead cytometry, fluorescence anisotropy and RT-qPCR experiments. Significance was set at p < 0.05 (*), p < 0.01(**) and p < 0.001 (***). For the proteomic study, after alignment and normalization, statistical analysis was performed using one-way analysis of variance (ANOVA) calculations. The significance of the peroxynitrite labelling was determined using two-way ANOVA calculations.
Results
NO2 exposure alters P. fluorescens MFAF76a cells morphology
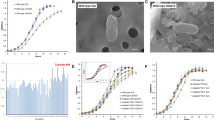
Previous work by T. Kondakova and coworkers determined that P. fluorescens MFAF76a showed significant modifications when exposed to 45 ppm of NO2, and we therefore chose to focus on this concentration21. To determine if NO2 exposure changes the cell morphology, P. fluorescens strain MFAF76a cells were observed by scanning electron microscopy after exposure to 45 ppm of NO2 (Fig. 1a). Around 35% of bacteria exposed to NO2 displayed impaired cell surface as compared to the control (Fig. 1a). The aspect of altered cell envelopes was rough and bumpy, presenting asperities and potential cavities. Flow cytometry assay analyses showed that P. fluorescens MFAF76a cells exposed to NO2 had a significant increase of the forward scatter (FSC), which indicates an augmentation of their size (Fig. 1b,c).

Morphology of P.fluorescens MFAF76a after exposure to NO2 45 ppm. (a) P. fluorescens MFAF76a strain observed by scanning electron microscopy. After exposure to either synthetic air (control) or 45 ppm of NO2 (NO2 45 ppm) for 2 h, P. fluorescens MFAF76a were fixed and observed by scanning electron microscopy, revealing envelope alterations (yellow arrows). (b) Forward scatter (FSC-A) curve of MFAF76a cells exposed to 45 ppm of NO2 (NO2 45 ppm) or synthetic air (Control). (c) Forward scatter (FSC-A) of the 1st, 5th and 9th deciles of MFAF76a cells exposed to 45 ppm of NO2 or synthetic air. Statistical significance was determined using a Mann–Whitney test (N = 5). n.s. = p > 0.05 * = p < 0.05.
NO2 exposure modifies the quantities of proteins involved in divisome formation as well as in the synthesis of envelope components
To get further insights into the potential causes of the NO2-induced alterations on cell morphology of P. fluorescens MFAF76a, a global proteomic profiling analysis was performed comparing NO2 or synthetic air exposure of bacteria. The proteomic data of MFAF76a has revealed the modulation of the quantities of several proteins involved in the formation of the components of the bacterial envelope and Z-ring formation (Fig. 2b).

Modifications in the proteomic profile of MFAF76a relative to envelope component synthesis and z-ring formation after exposure to 45 ppm of NO2. (a) Biosynthesis pathways of peptidoglycan and lipopolysaccharides. (b) Changes in the abundance of proteins involved in the synthesis and metabolism of various components of the bacterial envelope, as well as in the cell cycle, after exposure to 45 ppm of NO2. The proteins were identified using the P. fluorescens A506 strain as reference (N = 4). * = p < 0.05; ** = p < 0.01; *** = p < 0.001.
The proteomic study data revealed variations in the quantity of proteins involved in the synthesis of uridine diphosphate N-acetylglucosamine (UDP-GlcNAc), the precursor of peptidoglycan and lipopolysaccharides (Fig. 2a). These proteins are GlmS, GlmM and GlmU. The results also revealed decreased amounts of FtsI, DacA and MurC, all involved in peptidoglycan synthesis and cross-linking after exposure to NO2.
The proteome study analysis also revealed important amount modifications variations of the proteins MraZ, FtsZ, FtsE, FtsA, FtsI and MinE playing a role in the formation of the septal ring29.
NO2 exposure alters P. fluorescens MFAF76a membrane fluidity and integrity
Aside from peptidoglycan, the other critical components in the structure of the cell envelope are the cell membranes. To clarify whether the effects of NO2 observed by electronic microscopy and flow cytometry are also mediated by membrane alterations, membrane integrity and membrane fluidity were assessed for bacteria exposed to NO2 or synthetic air. Membrane integrity assays were performed using the green-fluorescent membrane-permeant DNA probe SYTO9™ and the red-fluorescent membrane impermeant DNA probe propidium iodine (PI). Their fluorescence was measured by flow cytometry (Fig. 3b). Three populations of cells were distinguishable after labelling (Fig. 3a). Cells with intact membranes were only labeled by SYTO9 ™, while cells with a damaged membrane were labelled by both PI and SYTO9™. However, cells labelled by PI did not form a single population. Some cells formed a highly homogenous population displaying high red fluorescence and a green fluorescence in the lower ranges of SYTO9™ labelled cells (damaged membranes, Fig. 3a). This profile was similar to the control cells permeabilized by 50% ethanol, suggesting that their membranes completely lost their integrity. However, another, less homogenous, population of cells was fluorescent in the red and displayed a higher and more heterogeneous green fluorescence (partially damaged membranes, Fig. 3a). This profile was not found in the permeabilized cell control, suggesting that the membranes of these cells were only partially damaged. The PI fluorescence threshold used to discriminate between the partially damaged and the intact cells was put at the maximum PI fluorescence detected in non-labelled cells, sometimes resulting in the splitting of this population between partially damaged membranes and intact membranes. The SYTO9™ fluorescence threshold to discriminate between partially and totally damaged membranes was determined thanks to the cells permeabilized by 50% ethanol. Interestingly, the results showed a dramatic increase of the number of cells labelled by PI upon exposure to NO2. Indeed, 42.5% of the cells exposed to NO2 were damaged, as compared to 2.5% of the control cells (Fig. 3b). Meanwhile, 18.2% of the exposed bacteria and 7.1% of the control bacteria (Fig. 3b) displayed the profile associated with partially damaged membranes. Membrane fluidity of P. fluorescens MFAF76a was determined by measuring the fluorescence anisotropy of the 1,6-Diphenyl-1,3,5-hexatriene (DPH) fluorescent probe (Fig. 3c). DPH is hydrophobic and localizes at the core of the phospholipid bilayer, at the level of the fatty acid chains30. Excitation of the probe by polarized light quantifies the polarization of emitted fluorescence. The polarization of emitted fluorescence depends on the capacity of the probe to rotate upon itself. Therefore, the fluorescence polarization is proportional to the fluidity of the lipid bilayer in which the probe is inserted31,32. DPH behavior can also be affected by its ratio to lipids, but previous gas chromatography analysis did not show any difference in terms of quantity, composition and cis/trans ratio of fatty acid between the two conditions33. Our results showed that fluorescence anisotropy was significantly reduced to 0.192 after bacterial exposure to NO2 (45 ppm) as compared to 0.217 for control condition (synthetic air), indicating an increased membrane fluidity (Fig. 3c).

Membrane fluidity and integrity of P. fluorescens MFAF76a upon exposure to NO2. Bacterial cells were exposed to 45 ppm of NO2 (NO2 45 ppm) or to synthetic air (Control) for 2 h. (a) Intensity of cell fluorescence in the PI and SYTO9 spectra of emission. (b) Membrane integrity assessed by live-dead flow cytometry assays using live-dead kit according to the manufacturer’s recommendations. (c) Membrane fluidity was determined by fluorescence anisotropy at 28 °C using DPH probe. Statistical significance was determined using t-test (N ≥ 4). ** = p < 0.01; *** = p < 0.001.
To clarify the consequences of the membrane alterations observed by cytometry and anisotropy, a proteomic profiling analysis of the proteins involved in the synthesis of membrane components was performed (Fig. 4). KdsC and LpxC, two proteins involved in the early steps of lipopolysaccharides biosynthesis were more detected after exposure to NO2. Furthermore, three proteins involved in the formation of the acetyl-CoA-carboxylase complex (ACC), responsible for the early step of fatty acid biosynthesis, were present in lower amounts in the condition exposed to NO2. These proteins are AccA, AccC and AccD. Furthermore, FabG and FabA, involved in latter steps of fatty acid biosynthesis were overproduced after NO2 exposure. Finally, FadE, involved in the first step of fatty acid metabolism through beta-oxidation was found in lower amounts compared to the control condition. However, proteins involved in phospholipid biosynthesis were not identified.

Modifications in the proteomic profile of MFAF76a relative to fatty acid and LPS biosynthesis and metabolism after exposure to 45 ppm of NO2. (a) Biosynthesis pathways of fatty acids in Pseudomonas spp. (b) Changes in the abundance of proteins involved in the synthesis and metabolism of fatty acids and lipopolysaccharide after exposure to 45 ppm of NO2. The proteins were identified using the P. fluorescens A506 strain as reference (N = 4). * = p < 0.05; ** = p < 0.01; *** = p < 0.001.
In a previous study, we showed slight modifications in the phospholipid composition of P. fluorescens MFAF76a membranes after exposure to NO2, including the disappearance of a minor unknown glycerophospholipid (UGP), but no lipid peroxidation seems to take place33. These previous results combined with the observed membrane alterations noted in Fig. 1 suggest that NO2 exposure could have an impact on the cell phospholipids. By searching the genome of MFAF76a strain, ten genes involved in the different steps of phospholipid biosynthesis from glycerol-3-phosphate (glycerol-3P) and acetyl-coenzyme A (acetyl-CoA) were found (cdsA, clsA, clsB, clsC, pgpA, pgsA, pssA, plsC, psd and pcs) (Fig. S1a). Since the proteins involved in phospholipid biosynthesis in P. fluorescens were not detected by the proteomic profiling analysis, the transcriptional regulation of the genes encoding for these proteins was assessed (Fig. S1b). RT-qPCR assays (Fig. S1b) showed an important transcription decrease of clsB, encoding the cardiolipin synthase B (ClsB). However, no significant expression levels difference was noted for the other studied genes upon NO2 exposure.
Discussion
NO2 is an important air pollutant whose effects on the human lung microbiota are poorly understood. To study the effects of atmospheric NO2 on bacteria, the airborne P. fluorescens MFAF76a strain was exposed to either 45 ppm of gaseous NO2 or synthetic air for 2 h, through an exposition system described previously21.
In this study, electronic microscopy observations revealed that exposure to 45 ppm of NO2 affects the envelope cell morphology of MFAF76a strain (Fig. 1a). Indeed, while some control cells displayed a rough cell surface, the prevalence and intensity of these irregularities were much higher among cells exposed to NO2. The effect of NO2 on the bacterial envelope is reminiscent of the study of Deupree and Schoenfisch that showed the deterioration of P. aeruginosa membranes by NO, and outlined even potential holes formation34, and suggests that NO2 could alter membranes in a similar fashion. In this study, we tried to explain the emergence of the observed phenotype and to understand the effects of NO2 on the various cell components.
Flow cytometry showed that exposure to NO2 induces an increase in cell size (Fig. 1b,c). However, the cell size did not increase homogeneously, as the cells exposed to NO2 displayed more high sized events (Fig. 1b,c). This increase of the number of cells of high size could be the result of an alteration of the cell division process. This alteration is however temporary as the generation time of the bacteria published in Depayras et al. were not different after exposure to NO233. The growth curve displayed a lag phase, which was at least partially explained by a significant diminution of cell viability.
To gain further insights into the observed cellular morphology modifications in MFAF76a strain, a proteome profiling approach was assessed in the absence or presence of NO2. Interestingly, the proteomic profiling data (Fig. 2b) revealed modifications of the amounts of several key proteins involved in the formation of the septal ring after exposure to NO2, supporting the results found by flow cytometry (Fig. 1b). Septal ring formation is a complex process. Briefly, FtsZ polymerizes into a z-ring at the midcell and recruits other proteins such as FtsA and ZipA that anchor the z-ring to the membrane. This complex then recruits other effector proteins that will help in the contraction of the ring and the synthesis of cell wall at the division site to prevent the rupture of the cell. Z-ring localization is regulated by the Min complex, which prevents the polymerization of FtsZ near the poles of the cell35. Among the proteins involved in the bacterial divisome formation, six were differentially expressed upon exposure to NO2. These proteins are FtsA, FtsE, FtsI, FtsZ, MraZ and MinE. The protein FtsZ can polymerize into a protofilament forming the z-ring, and allows the recruitment of other effector proteins. This polymerization is inhibited at one cell pole by the MinCD complex, which is then separated by MinE, causing MinCD to reform at the opposite pole, only to be separated by MinE again. This oscillation of MinCD and MinE restricts the polymerization of FtsZ to the center of the cell. FtsE is part of the transmembrane FtsEX complex, an ABC transporter-like protein required for cell division in low salt environment36. This complex is located to the septal ring through FtsE, while FtsX has multiple functions, including sequestration of FtsA to prevent its dimerization and recruitment of proteins involved in peptidoglycan hydrolysis37. The increase in FtsZ, MinE and FtsE quantities after NO2 exposure suggests the activation of regulatory pathways promoting cell division. However, another essential protein, FtsA, is under-expressed by threefold under nitrosative stress. FtsA is partly responsible for the attachment of the z-ring to the cell membrane as well as the recruitment of other downstream effector proteins necessary for cell constriction. Through its ability to bind FtsZ and the membrane, FtsA plays an important role in FtsZ dynamics. FtsA acts as a membrane anchor for FtsZ, but also tends to destabilize FtsZ polymers, and an overproduction of FtsA impairs cell division. Furthermore, FtsA is also able to self-polymerize which could inhibit its functions. This process could not be totally assumed since literature reports seemingly contradictory results on this subject37,38,39. Therefore, several hypotheses could explain the sharp decrease of the quantity of FtsA inside the cell after exposure to NO2. Lowering the production of FtsA could be a way for the cell to improve the septal ring stability by promoting FtsZ polymerization. FtsA could also be degraded after failing to attach to a membrane disorganized by NO2. Finally, FtsI abundance is also decreased by threefold. This protein is responsible for the synthesis of peptidoglycan at the septal ring location. This biosynthesis allows the contraction of the membrane without destroying the cell envelope. Normal localization of FtsI at the septum requires FtsA among other proteins. Since most proteins involved in the septal ring division are bound to the inner membrane, either directly or through their interaction with other proteins, perturbations of the membrane integrity could modulate their efficacy.
Furthermore, several proteins involved in the synthesis of peptidoglycan are differentially produced after exposure to NO2 (Fig. 2b). Thus, GlmS is overproduced while GlmM and GlmU are both underproduced in response to NO2 exposure. GlmS and GlmM reaction products are not specific to the peptidoglycan pathway, so GlmU decrease is a better indicator of the effect of NO2 regarding peptidoglycan synthesis. Peptidoglycan synthesis requires the presence of two precursors, uridine diphosphate N-acetylglucosamine (UDP-GlcNAc) and uridine diphosphate N-acetylglucosamine (UDP-Mur-NAc)40. Furthermore, MurC, involved in the addition of the pentapeptide moiety to UDP-Mur-NAc is also underproduced. Finally, FtsI, responsible for peptidoglycan cross-linking at the site of the division septum, and DacA, which mediates peptidoglycan cross-linking are less abundant41,42. These results show that peptidoglycan synthesis is underproduced upon exposure to NO2 through the limitation of the synthesis of its precursor. Furthermore, a decrease in DacA abundance can alter bacterial morphology, by producing more irregular cells43. The decreased DacA quantity after exposure to NO2 could therefore contribute to the cell surface irregularities observed by electron microscopy.
The morphological alterations could also be linked to modifications of the membranes. To ascertain if the cell surface and elongated cells phenotypes are associated to membrane integrity alterations, membrane integrity measurements were evaluated in MFAF76a strain in the absence or presence of NO2 (Fig. 3a, b). The integrity data showed that membranes of 42.5% of the cells exposed to NO2 were completely permeant to PI, while being only weakly labelled by SYTO9™. Meanwhile, 18.2% of the cells exposed to NO2 showed strong labelling with both PI and SYTO9™ (Fig. 3b). This co-labelling indicates that the cells where less permeant to PI, suggesting their membranes had a higher level of integrity than the cells entirely permeant to PI. These values, compared with the respective 2.5% and 7.1% of the control cells, suggest that the cell morphology alterations observed by electron microscopy could be correlated to a decrease of the membrane integrity. These results prompted us to also explore the membrane fluidity which was quantified by fluorescence anisotropy using the DPH probe (Fig. 3c). Exposure to NO2 decreased significantly DPH fluorescence anisotropy in MFAF76a, indicating an increase of membrane disorganization. This increase could be the result of alterations to the membrane phospholipids, as well as damages to membrane proteins caused by NO2 and its potential derivatives. Together, these results confirm that the membranes of the MFAF76a strain were strongly altered upon NO2 exposure.
The proteomic profiling analysis of proteins involved in the synthesis of membrane components revealed that these pathways are also affected by NO2 (Fig. 4b). UDP-GlcNAc, involved in an early step of PG synthesis, is also the precursor of lipid A, a part of lipopolysaccharide (LPS) when associated with a polysaccharide. The first stage of the LPS biosynthesis pathway is the conversion of UDP-GlcNAc into Kdo2-lipid A by nine enzymes44. The protein LpxC, which is strongly, overproduced after exposure to NO2, catalyses the first committed step of this conversion. Synthesis of Kdo2-lipid A also requires the addition of two CMP-3-deoxy-D-manno-octulosonic acid (CMP-Kdo) residues44. KdsC is the second of the three enzymes of the subpathway synthetizing CMP-Kdo from D-ribulose-5-phosphate45. This enzyme is also overproduced in cells exposed to NO2. Taken together, these results suggest that NO2 had an effect on synthesis of the peptidoglycan and lipopolysaccharides precursor UDP-GlcNAc.
Furthermore, the quantity of several proteins involved in fatty acid biosynthesis (FAB) was modulated in response to NO2 exposure (Fig. 4b). ACC is a four-subunit complex catalysing the first step of FAB in Pseudomonas46. Three of these subunits, AccA, AccC and AccD, were underproduced in the cells exposed to NO2, suggesting decreased FAB. Two proteins involved in the elongation of fatty acid, FabA and FabG, are overproduced after NO2 exposure (Fig. 4a). Taken together, these results indicate that NO2 exposure altered FAB pathway in MFAF76a strain. The overproduction of FabA could cause the increase of cis-unsaturated fatty acids, leading to an increase of membrane fluidity. However, the increase of membrane fluidity is unlikely to be caused by increase of cis-UFA production, since in a previous studies we showed that NO2 exposure did not induce any significant alteration of the degree of saturation nor in cis–trans isomerization ratio of fatty acids33. Moreover, FadE, which catalyzes the first step of fatty acid beta-oxidation, was underproduced after exposure to NO247, indicating that the beta-oxidation of fatty acids is also impacted by NO2 and may be downregulated. Interestingly, despite the modification observed in membrane components biosynthesis, the bacterial colonies displayed no observable differences between the control and the cells exposed to NO2.
The proteomic profiling study showed that gaseous NO2 have an important effect on proteins synthesizing most of the envelope components. However, abundance variations on proteins involved in phospholipid biosynthesis was not detected with this approach. To assess whether bacteria modulate their phospholipid biosynthesis in response to NO2, the transcription of genes encoding for enzymes involved in this process were analyzed by RT-qPCR. No significant differences in expression levels of genes involved in phospholipid biosynthesis between control cells and NO2 exposed cells were found except for clsB, the expression of which was reduced by eightfold. clsB encodes for the cardiolipin synthase B protein, involved in the last step of the cardiolipin biosynthesis pathway, and is only active in stationary phase of growth48. Despite the presence of three cardiolipin synthase genes in the MFAF76a strain, no cardiolipin was characterized in the lipidomic profile of the MFAF76a strain33. However, it is possible that the absence of cardiolipin could be due to the methods used to extract and analyze the cardiolipin profile. Cardiolipin is known to have an important role in membrane structure, by increasing membrane fluidity or by producing a favorable environment for the correct functioning of membrane proteins49. Cardiolipin is also known to be localized preferentially at the poles of the cell. Therefore it plays a role in determining the localization of proteins involved in the septal ring formation such as the Min complex by favoring its biding to the membrane at the cell poles50,51. These data show that exposure to NO2 affected the cardiolipin biosynthesis pathway, which might lead to a reconfiguration of the bacterial envelope, in accordance with UGP loss upon NO2 exposure.
Overall, these results show that NO2 exposure of MFAF76a leads to an augmentation of the cell size, which may be linked to divisome formation impairment. Furthermore, NO2 induced envelope alterations and decrease membrane integrity. These alterations can be partially explained by the modification of numerous components of the cell envelope, including peptidoglycan, lipid A and phospholipids. Further experiments on other bacterial strains would permit the distinction between the general effect of NO2 on bacteria and the effects that may be specific to this strain.
Data availability
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Skalska, K., Miller, J. S. & Ledakowicz, S. Trends in NOx abatement: A review. Sci. Total Environ. 408, 3976–3989 (2010).
Burney, S., Caulfield, J. L., Niles, J. C., Wishnok, J. S. & Tannenbaum, S. R. The chemistry of DNA damage from nitric oxide and peroxynitrite. Mutat. Res. 424, 37–49 (1999).
Bartesaghi, S. & Radi, R. Fundamentals on the biochemistry of peroxynitrite and protein tyrosine nitration. Redox Biol. 14, 618–625 (2018).
Möller, M. N., Li, Q., Lancaster, J. R. & Denicola, A. Acceleration of nitric oxide autoxidation and nitrosation by membranes. IUBMB Life 59, 243–248 (2007).
Pryor, W. A. & Lightsey, J. W. Mechanisms of nitrogen dioxide reactions: initiation of lipid peroxidation and the production of nitrous acid. Science 214, 435–437 (1981).
Science, G. Spatial distribution and temporal variation in ambient ozone and its associated NOx in the atmosphere of Jeddah City, Saudi Arabia. Aerosol Air Qual. Res. 13, 1712–1722 (2013).
Kumar, U., Prakash, A. & Jain, V. K. A photochemical modelling approach to investigate O3 sensitivity to NOx and VOCs in the urban atmosphere of Delhi. Aerosol Air Qual. Res. 8(2), 147–159 (2008).
Itano, Y. et al. Impact of NOx reduction on long-term ozone trends in an urban atmosphere. Sci. Total Environ. 379, 46–55 (2007).
WHO | Air quality guidelines - global update 2005. WHO http://www.who.int/phe/health_topics/outdoorair/outdoorair_aqg/en/.
INERIS - Dioxyde d’azote. https://substances.ineris.fr/fr/substance/cas/10102-44-0/2.
Fang, F. C. Antimicrobial reactive oxygen and nitrogen species: concepts and controversies. Nat. Rev. Microbiol. 2, 820–832 (2004).
Zumft, W. G. Cell biology and molecular basis of denitrification. Microbiol. Mol. Biol. Rev. 61, 533–616 (1997).
Greenberg, E. P. & Becker, G. E. Nitrous oxide as end product of denitrification by strains of fluorescent pseudomonads. Can. J. Microbiol. 23, 903–907 (1977).
Almeida, J. S., Reis, M. A. & Carrondo, M. J. Competition between nitrate and nitrite reduction in denitrification by Pseudomonas fluorescens. Biotechnol. Bioeng. 46, 476–484 (1995).
Grosser, M. R. et al. Genetic requirements for Staphylococcus aureus nitric oxide resistance and virulence. PLOS pathogens 14, e1006907 (2018).
Kim, S. & Lee, Y. H. Impact of small RNA RaoN on nitrosative-oxidative stress resistance and virulence of Salmonella enterica serovar Typhimurium. J Microbiol. https://doi.org/10.1007/s12275-020-0027-2 (2020).
Lee, A. G. Lipid–protein interactions in biological membranes: A structural perspective. Biochim. et Biophys. Acta BBABiomembr. 1612, 1–40 (2003).
Norimatsu, Y., Hasegawa, K., Shimizu, N. & Toyoshima, C. Protein–phospholipid interplay revealed with crystals of a calcium pump. Nature 545, 193–198 (2017).
Liu, X., Miller, M. J. S., Joshi, M. S., Thomas, D. D. & Lancaster, J. R. Accelerated reaction of nitric oxide with O2 within the hydrophobic interior of biological membranes. PNAS 95, 2175–2179 (1998).
Poc, C. et al. Airborne fluorescent pseudomonads : What potential for virulence?. Int. J. Curr. Microbiol. App. Sci 3, 708–722 (2014).
Kondakova, T. et al. Response to gaseous NO2 air pollutant of p fluorescens airborne strain MFAF76a and clinical strain MFN1032. Front. Microbiol. 7, 379 (2016).
Bergeau, D. et al. Unusual extracellular appendages deployed by the model strain Pseudomonas fluorescens C7R12. PLoS ONE 14, e0221025 (2019).
Schneider, C. A., Rasband, W. S. & Eliceiri, K. W. NIH image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675 (2012).
Clamens, T. et al. The aliphatic amidase AmiE is involved in regulation of Pseudomonas aeruginosa virulence. Sci. Rep. 7, 1–16 (2017).
Bouffartigues, E. et al. Transcription of the oprF gene of Pseudomonas aeruginosa Is dependent mainly on the SigX sigma factor and is sucrose induced. J. Bacteriol. 194, 4301–4311 (2012).
Gicquel, G. et al. The extra-cytoplasmic function sigma factor SigX modulates biofilm and virulence-related properties in Pseudomonas aeruginosa. PLoS ONE 8, e80407 (2013).
Guyard-Nicodème, M. et al. Outer membrane modifications of Pseudomonas fluorescens MF37 in response to hyperosmolarity. J. Proteome Res. 7, 1218–1225 (2008).
Bouteiller, M. et al. Crosstalk between the Type VI secretion system and the expression of class IV flagellar genes in the Pseudomonas fluorescens MFE01 Strain. Microorganisms 8, 622 (2020).
Wang, M., Fang, C., Ma, B., Luo, X. & Hou, Z. Regulation of cytokinesis: FtsZ and its accessory proteins. Curr. Genet. 66, 43–49 (2020).
Pebay-Peyroula, E., Dufourc, E. J. & Szabo, A. G. Location of diphenyl-hexatriene and trimethylammonium-diphenyl-hexatriene in dipalmitoylphosphatidylcholine bilayers by neutron diffraction. Biophys. Chem. 53, 45–56 (1994).
Lentz, B. R. Membrane, “fluidity” as detected by diphenylhexatriene probes. Chem. Phys. Lipid. 50, 171–190 (1989).
Lentz, B. R. Use of fluorescent probes to monitor molecular order and motions within liposome bilayers. Chem. Phys. Lipid. 64, 99–116 (1993).
Depayras, S. et al. Impact of gaseous NO2 on P. fluorescens strain in the membrane adaptation and virulence. Int. J. Environ. Impacts 1(2), 183–192 (2018).
Deupree, S. M. & Schoenfisch, M. H. Morphological analysis of the antimicrobial action of nitric oxide on Gram-negative pathogens using atomic force microscopy. Acta Biomater 5, 1405–1415 (2009).
Rowlett, V. W. & Margolin, W. The Min system and other nucleoid-independent regulators of Z ring positioning. Front. Microbiol. 6, 478 (2015).
Schmidt, K. L. et al. A predicted ABC transporter, FtsEX, Is needed for cell division in Escherichia coli. J Bacteriol 186, 785–793 (2004).
Du, S., Pichoff, S. & Lutkenhaus, J. FtsEX acts on FtsA to regulate divisome assembly and activity. Proc. Natl. Acad. Sci. U.S.A 113, E5052–E5061 (2016).
Krupka, M. et al. Role of the FtsA C terminus as a switch for polymerization and membrane association. MBio 5, e02221-14 (2014).
Nag, D., Chatterjee, A. & Chakrabarti, G. FtsA-FtsZ interaction in Vibrio cholerae causes conformational change of FtsA resulting in inhibition of ATP hydrolysis and polymerization. Int J Biol Macromol 142, 18–32 (2020).
Pazos, M. & Peters, K. Peptidoglycan. In Bacterial cell walls and membranes 127–168 (Springer, Cham, 2019). https://doi.org/10.1007/978-3-030-18768-2_5.
de Pedro, M. A., Quintela, J. C., Höltje, J. V. & Schwarz, H. Murein segregation in Escherichia coli. J. Bacteriol. 179, 2823–2834 (1997).
Yang, H. et al. Improving extracellular protein production in Escherichia coli by overexpressing D, D-carboxypeptidase to perturb peptidoglycan network synthesis and structure. Appl. Microbiol. Biotechnol. 103, 793–806 (2019).
Pan, X. et al. Loss of serine-type D-Ala-D-Ala carboxypeptidase DacA enhances prodigiosin production in Serratia marcescens. Front. Bioeng. Biotechnol. 7, 367 (2019).
Wang, X. & Quinn, P. J. Lipopolysaccharide: Biosynthetic pathway and structure modification. Prog. Lipid Res. 49, 97–107 (2010).
Wu, J. & Woodard, R. W. Escherichia coli YrbI Is 3-deoxy-d-manno-octulosonate 8-phosphate phosphatase. J. Biol. Chem. 278, 18117–18123 (2003).
Kondakova, T. et al. Glycerophospholipid synthesis and functions in Pseudomonas. Chem. Phys. Lipid. 190, 27–42 (2015).
Campbell, J. W. & Cronan, J. E. Jr. The enigmatic Escherichia coli fadE gene is yafH. J. Bacteriol. 184, 3759–3764 (2002).
Guo, D. & Tropp, B. E. A second Escherichia coli protein with CL synthase activity. Biochim. Biophys. Acta 1483, 263–274 (2000).
Unsay, J. D., Cosentino, K., Subburaj, Y. & García-Sáez, A. J. Cardiolipin effects on membrane structure and dynamics. Langmuir https://doi.org/10.1021/la402669z (2013).
Kawai, F. et al. Cardiolipin domains in Bacillus subtilis marburg membranes. J. Bacteriol. 186, 1475–1483 (2004).
Mileykovskaya, E. & Dowhan, W. Role of membrane lipids in bacterial division-site selection. Curr. Opin. Microbiol. 8, 135–142 (2005).
Acknowledgements
This work was financially supported by the GRR SESA (Région Normandie), and the European Union (European Regional Development Fund, ERDF), “Région Normandie” and “Évreux Portes de Normandie” (France). Proteomic experiments were cosupported by European Union and Région Normandie. Europe gets involved in Normandie with ERDF.
Author information
Authors and Affiliations
Contributions
Conceptualization, T.C., S.D. and C.D.-P.; methodology, T.C., S.D., D.S., T.K., M.B., J.H., A.T., O.T., Y.K.-G., C.B., T.K. and C.D.-P.; investigation, T.C., S.D., D.S. and C.D-P.; writing—original draft preparation, T.C., S.D., C.D.-P.; writing—review and editing, T.C., S.D., T.K., A.T., G.L., S.C., H.H., C.D.-P.; supervision, C.D.-P.; project administration, C.D.-P.; funding acquisition, N.O. and C.D.-P. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chautrand, T., Depayras, S., Souak, D. et al. Gaseous NO2 induces various envelope alterations in Pseudomonas fluorescens MFAF76a. Sci Rep 12, 8528 (2022). https://doi.org/10.1038/s41598-022-11606-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-11606-w
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
