


 Figure 1
Figure 1















« Prev Next »
It
seems nearly impossible for a normal cell to become a cancer cell unless it
inactivates the p53 network. Our current understanding of this network has come
from diverse lines of investigation, many initiated by serendipitous findings
that only later converged into a coherent picture. This story provides insights
into the nature of cancer, as well as the nature of scientific progress.
How Was p53 Discovered?
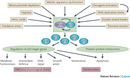
In 1979, six groups of investigators, each working independently, reported the discovery of a 53 kDa protein that was present in human and mouse cells (DeLeo et al. 1979, Kress et al. 1979, Lane & Crawford 1979, Linzer & Levine 1979, Melero et al. 1979, Smith et al. 1979). In five of these studies, the protein was discovered because it bound to the large T-antigen in SV40 infected cells and was therefore co-immunoprecipitated with antibodies generated against the viral protein. The same protein was discovered serendipitously when an antiserum generated against a chemically-induced mouse sarcoma was found to react with a 53 kDa protein present in transformed but not normal mouse cells (Figure 1).
Why Was p53 Originally Believed to Be an Oncogene?
A variety of studies carried out with the protein, and later with the gene encoding p53, indicated that it was an oncogene (Eliyahu et al. 1984, Jenkins et al. 1984, Parada et al. 1984, Eliyahu et al. 1985). This interpretation reflected both the research climate of the time and apparently compelling experimental evidence. Oncogenes were thought to be the key to understanding cancer, and had been identified in both RNA and DNA tumor viruses. In contrast, the existence of tumor suppressor genes was entirely conjectural, and barely on the radar of most cancer researchers. The p53 protein was bound to the major oncogenic protein of SV40, strongly suggesting that it was a downstream effector of the large T-antigen pathway. This interpretation was consistent with the high levels of expression of p53 found in many cancers. And the piece de resistance was the discovery that the introduction of a "normal" p53 gene into a normal cell could transform it, converting it into a tumor cell. Though there were a few experimental observations that did not fit well with the idea that p53 was an oncogene, there was little reason to believe otherwise in the mid 1980s.
How Do We Know p53 Is a Tumor Suppressor Gene?

© 2010 Nature Publishing Group Vogelstein, B. & Prives, C. The rise of p53. Nature Reviews Cancer (2009). All rights reserved. 
When this test was applied to p53, the results were entirely unanticipated. First, the majority of colorectal tumors were surprisingly found to have subtle mutations of p53, generally a single base substitution (such as C to T) resulting in a new amino acid (Baker et al. 1989). Mutations like this in p53 had never been observed before. Second, in virtually all cases, both copies of p53 were mutated. One copy was generally altered by a base substitution and the other copy was often completely deleted from the cell. This was the result expected for a tumor suppressor gene, not an oncogene. This "two-hit" test was then applied to many other tumor types and a similar result was found (Nigro et al. 1989). This result not only catapulted p53 into the center stage of human tumor research, but also provided cogent evidence that p53 was actually a tumor suppressor gene. This conclusion was confirmed by the subsequent findings that patients with inherited mutations of p53 were predisposed to diverse tumor types (Malkin et al. 1990, Srivastava et al. 1990) and that mice with engineered "knock-outs" of the p53 gene were also tumor prone (Donehower et al. 1992, Lowe et al. 1993)). Subsequent studies have demonstrated that p53 is more frequently mutated in human tumors than any other gene in the genome, and > 25,000 mutations have so far been reported (IARCTP53database).
What Effects Does p53 Have on Cell Growth?
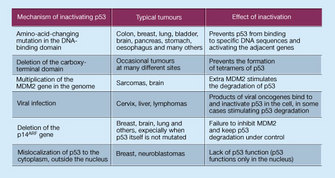
© 2000 Nature Publishing Group Vogelstein, B., Lane, D. & Levine, A. J. Surfing the p53 network. Nature 408, 307-310 (2000) doi:10.1038/35042675. All rights reserved.
In a classic study in 1991, wt p53 expression in leukemia cells resulted in their death by suicide (apoptosis) (Yonish-Rouach et al. 1991, Shaw et al. 1992). This observation proved a major stimulant to the burgeoning field of apoptosis and showed that the regulation of cell death was as important to tumor suppressor gene action as the regulation of cell birth. It is now understood that wt p53 regulates both aspects of net cell growth, inhibiting the cell cycle in some circumstances, while promoting apoptosis in others (Figure 2).
How Does p53 Work at the Biochemical Level?
In the early 1990s, two seminal observations were made: that p53 had a transactivation domain, and that it could bind to specific DNA sequences (Bargonetti et al. 1991, Kern et al. 1991). It was quickly demonstrated that the full length wt p53 protein functions as a sequence-specific transcriptional activator in vivo and in vitro (Farmer et al. 1992, Funk et al. 1992). Importantly, tumor derived mutants virtually always lost this critical function. Derivation of the first p53 consensus binding site revealed an unusually large and flexible sequence, with key invariant nucleotides (Bargonetti et al. 1991, el-Deiry et al. 1992). The crystal structure of the p53 core domain bound to DNA showed exactly which p53 residues made contact with DNA and revealed the way in which mutations abrogated this binding. Subsequent studies identified many genes that were transcriptionally activated by p53 through p53 binding sites in their regulatory regions. These included the cell cycle inhibitor CDKN128, and the pro-apoptotic proteins PUMA (Nakano & Vousden 2001, Yu et al. 2001) and NOXA (Oda et al. 2000), responsible in part for p53's control of cell growth.
What Regulates p53?
From the early days of p53 research, scientists knew that, in normal unstressed cells, p53 protein is scant, and that its turnover is rapid. Mdm2 was discovered in 1992 to bind to, and negatively regulate, transactivation by p53, and was then itself found to be a transcriptional target of p53, defining a negative feedback loop (Momand et al. 1992, Picksley & Lane 1993). Accordingly, the embryonic lethality of Mdm2 knockout mice could be rescued by knockout of p53 (de Rozieres et al. 2000). Later studies revealed a similarly important role for MdmX, an Mdm2 homolog (Shvarts et al. 1996). Mdm2 proved to be an E3 ubiquitin ligase, stimulating p53 degradation (Haupt et al. 1997, Honda et al. 1997, Kubbutat et al. 1997). It is now recognized that the action of ubiquitinases and deubiquitinases determines the activity of the p53 network. These findings explain the relatively high levels of p53 in tumors, as p53 mutants are transcriptionally inert, disrupting the feedback loop.
Is p53 Chemically Modified in the Cell?
The p53 protein is not active in the cell unless it is first modified by other proteins. In other words, the actual mass of p53 is not as important as the amount of activated p53, and only activated p53 can bind to DNA and stimulate the expression of its target genes. Although it was known since the 1980's that p53 levels increase after irradiation, only in 1992 was p53 shown to be regulated by ATM; a kinase orchestrating the DNA damage response (Banin et al. 1998, Canman et al. 1998). The p53 protein was subsequently demonstrated to be phosphorylated after DNA damage, and was the first non-histone protein shown to be acetylated by p300/CBP (Ionov et al. 2004). Biochemical studies showed DNA damage inducible kinases such as ATM (Westphal 1997) and Chk2 (Tominaga et al. 1999, Shieh et al. 2000) can phosphorylate key p53 residues that regulate its binding to Mdm2 and p300/CBP. The alternative reading frame (ARF) protein, known to be induced by a number of mitogens, was also shown to block the ability of Mdm2 to degrade p53, thereby linking p53 to key oncogenic pathways (Kamijo et al. 1998). The p53 protein has been shown to bind to dozens of other proteins, explaining its involvement in a wide range of physiologic processes.
What is the Future of p53 Research?
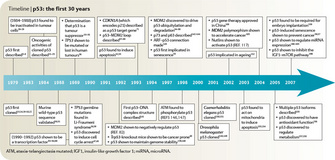
© 2009 Nature Publishing Group Levine, A. J. & Oren, M. The first 30 years of p53: growing ever more complex. Nature Reviews Cancer 9, 749-758 (2009). All rights reserved.
And perhaps most importantly, we don't yet know how to use the immense amount of knowledge so far gained about p53 for therapeutic purposes. Clever approaches to achieve this goal — small molecular weight compounds or peptides that reactivate mutant p53 or disrupt the interactions between MDM2 and wt p53, or viruses that only replicate in cells without a functional p53 network — have been developed and show great promise. However, the field is wide open to new, creative approaches that target p53, a protein that is inactivated in the majority of human cancers. History shows that the most novel ideas — the really bold and creative ones — often come from students.
Summary
The study of p53 has revealed many of the principles underlying human tumorigenesis. These include the critical differences between an oncogene and a tumor suppressor gene, the relationship between environmental exposures and cancer, the mechanisms through which cancer genes stimulate cell birth or inhibit cell death, and the striking networks that control the transcription, translation, and function of key cellular proteins. The many facets of these studies, coupled with the fact that p53 inactivation is essential for the formation of the majority of human tumors, has made p53 a uniquely valuable target for basic as well as applied research.
References and Recommended Reading
Baker, S. J., Fearon, E. R., et al. Chromosome 17 deletions and p53 gene mutations in colorectal carcinomas. Science 244, 217–221 (1989).
Banin, S., Moyal, L., et al. Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science 281, 1674–1677 (1998).
Bargonetti, J., Friedman, P. N., et al. Wild-type but not mutant p53 immunopurified proteins bind to sequences adjacent to the SV40 origin of replication. Cell 65, 1083–1091 (1991).
Canman, C. E., Lim, D. S., et al. Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science 281, 1677–1679 (1998).
DeLeo, A. B., Jay, G., et al. Detection of a transformation-related antigen in chemically induced sarcomas and other transformed cells of the mouse. Proceedings of the National Academy of Science, USA 76, 2420–2424 (1979).
de Rozieres, S., Maya, R., et al. The loss of mdm2 induces p53-mediated apoptosis. Oncogene 19, 1691–1697 (2000).
Donehower, L. A., Harvey, M., et al. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 356, 215–221 (1992).
Eliyahu, D., Raz, A., et al. Participation of p53 cellular tumour antigen in transformation of normal embryonic cells. Nature 312, 646–649 (1984).
Eliyahu, D., Michalovitz, D., et al. Overproduction of p53 antigen makes established cells highly tumorigenic. Nature 316, 158–160 (1985).
Eliyahu, D., Michalovitz, D., et al. Wild-type p53 can inhibit oncogene-mediated focus formation. Proceedings of the National Academy of Science, USA 86, 8763–8767 (1989).
el-Deiry, W. S., Kern, S. E., et al. Definition of a consensus binding site for p53. Nature Genetics 1, 45–49 (1992).
el-Deiry, W. S., Tokino, T., et al. WAF1, a potential mediator of p53 tumor suppression. Cell 75, 817–825 (1993).
Farmer, G., Bargonetti, J., et al. Wild-type p53 activates transcription in vitro. Nature 358, 83–86 (1992).
Finlay, C. A., Hinds, P. W. et al. The p53 proto-oncogene can act as a suppressor of transformation. Cell 57, 1083–1093 (1989).
Funk, W. D., Pak, D. T., et al. A transcriptionally active DNA-binding site for human p53 protein complexes. Molecular Cell Biology 12, 2866–2871 (1992).
Haupt, Y., Maya, R., et al. Mdm2 promotes the rapid degradation of p53. Nature 387, 296–299 (1997).
Honda, R., Tanaka, H., et al. Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS Letters 420, 25–27 (1997).
IARCTP53database .
Ionov, Y., Matsui, S., et al. A role for p300/CREB binding protein genes in promoting cancer progression in colon cancer cell lines with microsatellite instability. Proceedings of the National Academy of Science, USA 101, 1273–1278 (2004).
Jenkins, J. R., Rudge, K. et al. Cellular immortalization by a cDNA clone encoding the transformation-associated phosphoprotein p53. Nature 312, 651–654 (1984).
Kamijo, T., Weber, J. D., et al. Functional and physical interactions of the ARF tumor suppressor with p53 and Mdm2. Proceedings of the National Academy of Science, USA 95, 8292–8297 (1998).
Kern, S. E., Kinzler, K. W., et al. Identification of p53 as a sequence-specific DNA-binding protein. Science 252, 1708–1711 (1991).
Knudson, A.G., Jr. Mutation and cancer: statistical study of retinoblastoma. Proceedings of the National Academy of Science, USA 68, 820–823 (1971).
Kress, M., May, E., et al. Simian virus 40-transformed cells express new species of proteins precipitable by anti-simian virus 40 tumor serum. Journal of Virology 31, 472–483 (1979).
Kubbutat, M. H., Jones, S. N., et al. Regulation of p53 stability by Mdm2. Nature 387, 299–303 (1997).
Lane, D. P. & Crawford, L. V. T antigen is bound to a host protein in SV40-transformed cells. Nature 278, 261–263 (1979).
Linzer, D. I. & Levine, A. J. Characterization of a 54K dalton cellular SV40 tumor antigen present in SV40-transformed cells and uninfected embryonal carcinoma cells. Cell 17, 43–52 (1979).
Lowe, S. W., Schmitt, E. M., et al. p53 is required for radiation-induced apoptosis in mouse thymocytes. Nature 362, 847–849 (1993).
Malkin, D., Li, F. P., et al. Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science 250, 1233–1238 (1990).
Melero, J. A., Stitt, D. T., et al. Identification of new polypeptide species (48-55K) immunoprecipitable by antiserum to purified large T antigen and present in SV40-infected and -transformed cells. Virology 93, 466–480 (1979).
Momand, J., Zambetti, G. P., et al. The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell 69, 1237–1245 (1992).
Nakano, K. & Vousden, K. H. PUMA, a novel proapoptotic gene, is induced by p53. Molecular Cell 7, 683–694 (2001).
Nigro, J. M., Baker, S.J., et al. Mutations in the p53 gene occur in diverse human tumour types. Nature 342, 705–708 (1989).
Oda, E., Ohki, R., et al. Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science 288, 1053–1058 (2000).
Parada, L. F., Land, H., et al. Cooperation between gene encoding p53 tumour antigen and ras in cellular transformation. Nature 312, 649–651 (1984).
Picksley, S. M. & Lane, D. P. The p53-mdm2 autoregulatory feedback loop: a paradigm for the regulation of growth control by p53? Bioessays 15, 689–690 (1993).
Shaw, P., Bovey, R., et al. Induction of apoptosis by wild-type p53 in a human colon tumor-derived cell line. Proceedings of the National Academy of Science, USA 89, 4495–4499 (1992).
Shieh, S. Y., Ahn, J., et al. The human homologs of checkpoint kinases Chk1 and Cds1 (Chk2) phosphorylate p53 at multiple DNA damage-inducible sites. Genes and Development 14, 289–300 (2000).
Shvarts, A., Steegenga, W. T., et al. MDMX: a novel p53-binding protein with some functional properties of MDM2. The EMBO Journal 15, 5349–5357 (1996).
Smith, A.E., Smith, R., et al. Characterization of different tumor antigens present in cells transformed by simian virus 40. Cell 18, 335–346 (1979).
Srivastava, S., Zou, Z. Q., et al. Germ-line transmission of a mutated p53 gene in a cancer-prone family with Li-Fraumeni syndrome. Nature 348, 747–749 (1990).
Tominaga, K., Morisaki, H., et al. Role of human Cds1 (Chk2) kinase in DNA damage checkpoint and its regulation by p53. Journal of Biological Chemistry 274, 31463–31467 (1999).
Westphal, C. H. Cell-cycle signaling: Atm displays its many talents. Current Biology 7, R789–792 (1997).
Yonish-Rouach, E., Resnitzky, D., et al. Wild-type p53 induces apoptosis of myeloid leukaemic cells that is inhibited by interleukin-6. Nature 352, 345–347 (1991).
Yu, J., Zhang, L., et al. PUMA induces the rapid apoptosis of colorectal cancer cells. Molecular Cell 7, 673–682 (2001).










