
Featured
-
-
Article |
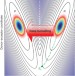 Wide-dynamic-range kinetic investigations of deep proton tunnelling in proteins
Wide-dynamic-range kinetic investigations of deep proton tunnelling in proteins
A temperature-dependent kinetic study of ground-state proton transfer in the green fluorescent protein highlights the role of ‘deep tunnelling’ in proton wires. A potential mechanism for directional proton transport is proposed, where high-pKa amino acid residues act as ‘tunnel diodes’ and as stabilizing elements within protein water wires.
- Bridget Salna
- , Abdelkrim Benabbas
- & Paul M. Champion
-
Article |
 The structural and chemical origin of the oxygen redox activity in layered and cation-disordered Li-excess cathode materials
The structural and chemical origin of the oxygen redox activity in layered and cation-disordered Li-excess cathode materials
The chemistry of the transition metals within the oxide cathodes of lithium-ion batteries typically limits their capacity, however, reversible oxygen redox could potentially break this limit. It is now demonstrated that Li-excess and cation disorder create specific environments around oxygen atoms that lead to labile oxygen electrons that participate in the practical capacity of cathodes.
- Dong-Hwa Seo
- , Jinhyuk Lee
- & Gerbrand Ceder
-
Article |
 Synthesis and stability of xenon oxides Xe2O5 and Xe3O2 under pressure
Synthesis and stability of xenon oxides Xe2O5 and Xe3O2 under pressure
The reactivity of the noble gases—a notoriously inert group—at high pressures is intriguing. Now, two xenon oxides with unusual stoichiometries, Xe2O5 and Xe3O2, have been synthesized above 78 GPa and predicted to be stable above 50 GPa, indicating that xenon is more reactive than previously thought.
- Agnès Dewaele
- , Nicholas Worth
- & Tetsuo Irifune
-
News & Views |
 Catch the carbon dioxide
Catch the carbon dioxide
Understanding the minute details of CO2 transport is key to finding new technologies that reduce the hazardous levels of CO2 in our atmosphere. Now, the observation that the transport of CO2 in molten calcium carbonate occurs faster than standard molecular diffusion brings us one step closer.
- Barbara Kirchner
- & Barbara Intemann
-
Article |
 Unimolecular dissociation dynamics of vibrationally activated CH3CHOO Criegee intermediates to OH radical products
Unimolecular dissociation dynamics of vibrationally activated CH3CHOO Criegee intermediates to OH radical products
An important source of atmospheric hydroxyl radicals is from the dissociation of Criegee intermediates produced in alkene ozonolysis reactions. The dissociation dynamics of the prototypical CH3CHOO Criegee intermediate have now been determined. Complementary experimental and theoretical studies were carried out and the translational and internal energy distributions of the OH radical products were characterized.
- Nathanael M. Kidwell
- , Hongwei Li
- & Marsha I. Lester
-
Article |
 Discovery of a regioselectivity switch in nitrating P450s guided by molecular dynamics simulations and Markov models
Discovery of a regioselectivity switch in nitrating P450s guided by molecular dynamics simulations and Markov models
A collaborative approach between experiment and simulation has revealed a single mutation in the F/G loop of the newly described nitrating cytochrome P450 TxtE that controls loop dynamics and, more surprisingly, the regioselectivity of the reaction. This mutation is present in a subset of homologous nitrating P450s that produce a previously unidentified biosynthetic intermediate, 5-nitro-L-tryptophan.
- Sheel C. Dodani
- , Gert Kiss
- & Frances H. Arnold
-
Article |
 Carbon dioxide transport in molten calcium carbonate occurs through an oxo-Grotthuss mechanism via a pyrocarbonate anion
Carbon dioxide transport in molten calcium carbonate occurs through an oxo-Grotthuss mechanism via a pyrocarbonate anion
The solvation behaviour of CO2 in carbonate melts is important from both a geochemical point of view and with respect to its electroreduction. Now, simulations have shown that solvation of CO2 in molten CaCO3 leads to the formation of the pyrocarbonate anion, C2O52–, which significantly enhances the transport of CO2 via a Grotthuss-like mechanism.
- Dario Corradini
- , François-Xavier Coudert
- & Rodolphe Vuilleumier
-
Article |
 Influence of the leaving group on the dynamics of a gas-phase SN2 reaction
Influence of the leaving group on the dynamics of a gas-phase SN2 reaction
Little is known about how the identity of a leaving group affects the dynamics of a bimolecular nucleophilic substitution reaction. A study of the reaction of F− with CH3Cl, and comparison to its reaction with CH3I, now reveals key insights into such effects, with reactant orientation considered a key factor in understanding the behaviour observed.
- Martin Stei
- , Eduardo Carrascosa
- & Roland Wester
-
Article |
 Unidirectional rotary motion in achiral molecular motors
Unidirectional rotary motion in achiral molecular motors
Avoiding equal probability for clockwise and anticlockwise rotation is essential for the function of molecular motors, and both biological and synthetic systems take advantage of chirality to control the rotary direction. Now it has been shown, by integrating two rotor moieties in a symmetric meso motor design, that light-driven unidirectional rotary motion can be achieved in an achiral system.
- Jos C. M. Kistemaker
- , Peter Štacko
- & Ben L. Feringa
-
Article |
 Molecular hydrogen interacts more strongly when rotationally excited at low temperatures leading to faster reactions
Molecular hydrogen interacts more strongly when rotationally excited at low temperatures leading to faster reactions
The rotational state of a molecule is not generally considered to play a role in how fast it reacts; however, when the temperature is low quantum effects become more important. Now, it is shown that at low temperatures rotationally excited H2 molecules react with He faster than non-rotating ground-state molecules — a process mediated by stronger long-range attraction.
- Yuval Shagam
- , Ayelet Klein
- & Edvardas Narevicius
-
Article |
 How do metal ions direct ribozyme folding?
How do metal ions direct ribozyme folding?
The question of how divalent metal ions direct the folding of ribozymes is a major unsolved problem. A computational model has now been used to reveal the molecular mechanism by which Mg2+ drives the Azoarcus ribozyme into a catalytically functional state. Simulations also show that although Ca2+ drives folding it leaves the active site unstable.
- Natalia A. Denesyuk
- & D. Thirumalai
-
Article |
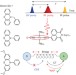 On the mechanism of vibrational control of light-induced charge transfer in donor–bridge–acceptor assemblies
On the mechanism of vibrational control of light-induced charge transfer in donor–bridge–acceptor assemblies
The ultrafast and mode-specific infrared excitation of several donor–bridge–acceptor (DBA) assemblies in solution has been shown to modulate their light-induced electron transfer properties. New insights are afforded into the role of vibrational processes immediately following light absorption in charge-transfer molecules and a recipe for efficient ‘vibrational control’ of electron transfer is proposed.
- Milan Delor
- , Theo Keane
- & Julia A. Weinstein
-
Article |
 Quantum interference between H + D2 quasiclassical reaction mechanisms
Quantum interference between H + D2 quasiclassical reaction mechanisms
Often reactions can be described by classical mechanics; however, this is prohibited in cases in which quantum phenomena emerge. Now angular distributions measured for the H + D2 reaction have been seen to display characteristic oscillation patterns in backward scattering — theory shows that they are caused by quantum interferences between classical mechanisms similar to those found in the double-slit experiment.
- Pablo G. Jambrina
- , Diego Herráez-Aguilar
- & Richard N. Zare
-
News & Views |
 Making a bad calculation
Making a bad calculation
Computations of the energetics and mechanism of the Morita–Baylis–Hillman reaction are “not even wrong” when compared with experiments. While computational abstinence may be the purest way to calculate challenging reaction mechanisms, taking prophylactic measures to avoid regrettable outcomes may be more realistic.
- Arthur Winter
-
-
Feature |
 Quantum reform
Quantum reform
Quantum computers potentially offer a faster way to calculate chemical properties, but the exact implications of this speed-up have only become clear over the last year. The first quantum computers are likely to enable calculations that cannot be performed classically, which might reform quantum chemistry — but we should not expect a revolution.
- Leonie Mueck
-
News & Views |
 A light-switched yin and yang pair
A light-switched yin and yang pair
In 1972, Baird showed theoretically that the electron counting rule for aromaticity and antiaromaticity in the lowest ππ* triplet state is opposite to that in the electronic ground state. A pair of compounds that manifests this reversal in character has now been identified and characterized experimentally for the first time.
- Henrik Ottosson
- & K. Eszter Borbas
-
News & Views |
 The quest for new functionality
The quest for new functionality
Building on our understanding of the chemical bond, advances in synthetic chemistry, and large-scale computation, materials design has now become a reality. From a pool of 400 unknown compositions, 15 new compounds have been realized that adopt the predicted structures and properties.
- Aron Walsh
-
Article |
 Prediction and accelerated laboratory discovery of previously unknown 18-electron ABX compounds
Prediction and accelerated laboratory discovery of previously unknown 18-electron ABX compounds
A method to predict the stability, structure and properties of as-yet-unreported materials has been devised. For 18-valence electron ABX materials, 15 such ‘missing’ compounds identified to be thermodynamically stable were successfully synthesized, and showed crystal structures and properties in good agreement with the predicted ones.
- Romain Gautier
- , Xiuwen Zhang
- & Alex Zunger
-
Correspondence |
Reply to 'Entropic factors also contribute to the high melting points of polyhedral alkanes'
- Sason Shaik
- & Santiago Alvarez
-
News & Views |
 Searching sequence space
Searching sequence space
Short peptides are among the most intriguing building blocks in nanotechnology, but it would be very challenging to experimentally study the properties of large numbers of different sequences. Now, a computational analysis of all 8,000 possible tripeptides has been used to identify those with interesting self-assembly behaviour.
- Ehud Gazit
-
Article |
 Exploring the sequence space for (tri-)peptide self-assembly to design and discover new hydrogels
Exploring the sequence space for (tri-)peptide self-assembly to design and discover new hydrogels
Peptides that self-assemble into nanostructures are of interest for many applications, including ones relevant to cosmetics, food, biomedicine and nanotechnology. Now, computational tools have been developed that enable peptide sequence space to be rapidly searched for supramolecular properties and this approach has been used to identify unprotected tripeptide hydrogelators.
- Pim W. J. M. Frederix
- , Gary G. Scott
- & Tell Tuttle
-
News & Views |
 A virtual squeeze on chemistry
A virtual squeeze on chemistry
Molecular simulations have the potential to give valuable insights into experimental results, but can be limited by the time- and length-scales they can simulate. Now, reactive chemistry can be driven through a novel simulation approach, which could have ramifications for many research areas, including astrobiology and the origins of life.
- Nir Goldman
-
Article |
 Discovering chemistry with an ab initio nanoreactor
Discovering chemistry with an ab initio nanoreactor
Computational chemistry is traditionally used to interpret experimental findings. Now its use in reaction discovery is described with the development of the ab initio nanoreactor — a highly accelerated, first-principles molecular dynamics simulation of chemical reactions that discovers new molecules and mechanisms without preordained reaction coordinates or elementary steps.
- Lee-Ping Wang
- , Alexey Titov
- & Todd J. Martínez
-
Article |
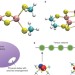 Low-energy spectrum of iron–sulfur clusters directly from many-particle quantum mechanics
Low-energy spectrum of iron–sulfur clusters directly from many-particle quantum mechanics
FeS clusters are a universal motif in organisms and are central to many processes, including nitrogen fixation and respiration. By carrying out the first many-electron calculation of the [2Fe-2S] and [4Fe-4S] clusters, they are shown to have an unusual set of closely packed energy levels, which are key to understanding their reactivity.
- Sandeep Sharma
- , Kantharuban Sivalingam
- & Garnet Kin-Lic Chan
-
-
Article |
 Direct observation of the collapse of the delocalized excess electron in water
Direct observation of the collapse of the delocalized excess electron in water
It is generally believed that, after being generated, an excess electron in water shrinks from a strongly delocalized to a localized state in about a picosecond. Now, these early stages in the behaviour of this electron have been observed using a combination of transient THz spectroscopy and ab initio molecular dynamics simulations.
- Janne Savolainen
- , Frank Uhlig
- & Pavel Jungwirth
-
-
Article |
 Incorporation of protein flexibility and conformational energy penalties in docking screens to improve ligand discovery
Incorporation of protein flexibility and conformational energy penalties in docking screens to improve ligand discovery
The adoption of multiple conformations by proteins presents a challenge for ligand discovery using docking simulations. Now, a method for representing the conformational behaviour of a flexible protein in docking screens, which is guided by experimental crystallography data, is shown to predict protein conformation, ligand pose and aid the discovery of new ligands.
- Marcus Fischer
- , Ryan G. Coleman
- & Brian K. Shoichet
-
Article |
 A transferable model for singlet-fission kinetics
A transferable model for singlet-fission kinetics
Understanding the process of exciton fission, which occurs in certain organic materials, could lead to the development of more efficient photovoltaic devices. Here, an expression derived from first principles is used to accurately characterize the singlet fission rate of a wide array of materials, reproducing a transition from weak to strong coupling as a function of molecular separation.
- Shane R. Yost
- , Jiye Lee
- & Troy Van Voorhis
-
News & Views |
 Isotope effects feel the cold
Isotope effects feel the cold
Kinetic isotope effects are widely used to elucidate reaction mechanisms and are generally interpreted in terms of simple kinetic models. Measurements of this effect for the Penning ionization reaction between helium and dihydrogen highlight the need for a quantum description of chemical reaction rates when sub-kelvin temperatures are approached.
- Mark Brouard
-
Article |
 Observation of the isotope effect in sub-kelvin reactions
Observation of the isotope effect in sub-kelvin reactions
In cold chemistry, quantum phenomena in reactants' translational motion lead to the temporary trapping of reactants in a collisional complex. It is now shown that this metastable complex is responsible for a dramatic quantum kinetic isotope effect as observed in Penning ionization reactions at low temperatures.
- Etay Lavert-Ofir
- , Yuval Shagam
- & Edvardas Narevicius
-
News & Views |
 It's all downhill from here
It's all downhill from here
High selectivity is essential in the enzymatic biosynthesis of complex natural products. Now, the discovery of multiple sequential bifurcations on the reaction path towards the formation of a diterpenoid shows how dynamics affect selectivity, and suggests how enzymes may steer reactions towards a specific product.
- Charles E. Hornsby
- & Robert S. Paton
-
Article |
 Biosynthetic consequences of multiple sequential post-transition-state bifurcations
Biosynthetic consequences of multiple sequential post-transition-state bifurcations
A terpene-forming carbocation reaction is described for which a single transition-state structure leads to the formation of many isomeric products via pathways that feature multiple sequential bifurcations. Dynamic effects are shown to contribute to the selectivity of the reaction, with consequences for how enzymes control the biosynthesis of complex natural products.
- Young Joo Hong
- & Dean J. Tantillo
-
-
-
News & Views |
 A stitch in time
A stitch in time
Lengthy molecular dynamics simulations of complex systems at the atomic scale usually require supercomputers. Now, by stitching together many shorter independent simulations run 'in the cloud', this requirement has been circumvented, allowing two milliseconds of the dynamics of a G-protein-coupled receptor to be simulated.
- Xavier Deupi
-
Article |
 Calculations predict a stable molecular crystal of N8
Calculations predict a stable molecular crystal of N8
Polynitrogen compounds are of interest on a fundamental level and as potential high-energy-density materials. A crystalline solid that consists of two isomeric forms of N8 molecules held together by weak van der Waals interactions has now been predicted to exist, and to be stable even at low pressures.
- Barak Hirshberg
- , R. Benny Gerber
- & Anna I. Krylov
-
Article |
 Cloud-based simulations on Google Exacycle reveal ligand modulation of GPCR activation pathways
Cloud-based simulations on Google Exacycle reveal ligand modulation of GPCR activation pathways
Two milliseconds of molecular dynamics simulations of a major drug-target G-protein-coupled receptor (GPCR) has been carried out using Google's Exacycle cloud computing platform. Markov state models were used to aggregate independent simulations into a statistical model that provides an atomistic description of GPCR ligand-modulated activation pathways.
- Kai J. Kohlhoff
- , Diwakar Shukla
- & Vijay S. Pande
-
-
-
-
-
News & Views |
 Beyond state I
Beyond state I
Although caesium is well known in its oxidation state +I, many chemists have speculated about a possible higher state. Such a species has not yet been prepared, but based on quantum-chemical calculations CsFn compounds have now been predicted to be stable.
- Sebastian Riedel
- & Peter Schwerdtfeger
-
News & Views |
 A bright future for defects
A bright future for defects
Covalently bonding groups to the walls of carbon nanotubes has been previously observed to quench their photoluminescence. Now, it has been shown that, if you get the chemistry just right, their photoluminescence can in fact be significantly brightened by introducing defects through functionalization.
- Qing Hua Wang
- & Michael S. Strano
-
Article |
 Caesium in high oxidation states and as a p-block element
Caesium in high oxidation states and as a p-block element
Caesium has so far not been found in oxidation states higher than +1, but quantum chemical calculations have now shown that, under high pressures, 5p inner shell electrons of caesium can participate in — and become the main components of — bonds. Caesium is predicted to form stable CsFn molecules that resemble isoelectronic XeFn.
- Mao-sheng Miao
-
News & Views |
 Inverse solvent design
Inverse solvent design
Choosing a solvent for a particular reaction is often a matter of personal preference or the result of limited screening. Now, a computational method allows identification of a solvent that will enhance the kinetics of a reaction prior to running a wet experiment.
- Donald G. Truhlar
-
-
News & Views |
 A multitude of spins
A multitude of spins
Accurately representing molecules with many coupled unpaired electrons is currently impossible using conventional electronic-structure theories. Now, using a recently developed approach, the near-exact quantum wavefunction of the highly complex Mn4CaO5 cluster of photosystem II has been calculated.
- Jeremy N. Harvey
 Accurate first-principles structures and energies of diversely bonded systems from an efficient density functional
Accurate first-principles structures and energies of diversely bonded systems from an efficient density functional Electrides explained
Electrides explained Molecular doormen
Molecular doormen Reduction to practice
Reduction to practice Cage calculations
Cage calculations Model citizens
Model citizens United they are strong
United they are strong State oddity
State oddity