
Abstract
Bats are efficient reservoirs of a number of viruses with zoonotic potential, and are involved directly in the transmission cycle of many zoonoses. In the present study, which is part of a larger project that is documenting the viromes of the bat species found in the Mid-North states of Maranhão and Piauí, we analyzed 16 pooled samples obtained from four species of bat of the genus Artibeus—Artibeus obscurus, Artibeus cinereus, Artibeus lituratus and Artibeus planirostris. We describe and identify a Hepatovirus, denominated Hepatovirus H isolate sotense, which was found in a pool of internal organs (liver and lungs) extracted from a specimen of A. planirostris, a frugivorous bat, collected in the Cerrado biome of Maranhão state. This material was analyzed using new generation sequencing, which produced a contig of 7390 nucleotides and presented a degree of identity with a number of existing Hepatovirus sequences available for bats (amino acid identity of 61.5% with Bat hepatovirus C of Miniopterus cf. manavi, 66.6% with Bat hepatovirus G of Coleura afra, 67.4% with Hepatovirus G2 of Rhinolophus landeri, and 75.3% with Hepatovirus H2 of Rhinolophus landeri). The analysis of the functional domains of this contig confirmed a pattern consistent with the characteristics of the genus Hepatovirus (Picornaviridae). In the phylogenetic tree with several other Hepatovirus species, this genome also grouped in a monophyletic clade with Hepatovirus H (HepV-H1; HepV-H2, and HepV-H3) albeit on an external branch, which suggests that it may be a distinct genotype within this species. This is the first isolate of Hepatovirus H identified in bats from South America, and represents an important discovery, given that most studies of viruses associated with bats in the state of Maranhão have focused on the family Rhabdoviridae.
Similar content being viewed by others
Bats are placental mammals of the order Chiroptera, with at least 1384 recognized species1, of which, approximately 15% occur in Brazil2. In addition to their ecological importance3, bats are also considered to be prominent virus hosts4. Pandemics caused by zoonoses reinforce the need for the monitoring of new viruses found in wild hosts, in particular, in bats and rodents, which are the mammals that harbor the greatest diversity of potentially zoonotic viral species5.
A total of 17 viral families have been identified in Brazilian bats, although only a small proportion (23.75%) have been the focus of research over the past 20 years6. The order Picornavirales—Pisoniviricetes: Pisuviricota7 is considered to be the largest and most diverse of the positive strand RNA viruses. The family Picornaviridae includes 158 species distributed in 68 genera8, which are capable of infecting an enormous range of vertebrate hosts. These viruses include some important pathogens, such as the poliovirus, and the viruses that cause hepatitis A and foot-and-mouth disease9.
A number of distantly-related hepatotropic viruses have recently been discovered in bats, rodents, and other small mammals10. These viruses are related phylogenetically, sharing antigenic determinants with the human hepatitis virus A11. Bats are known to have played an important role in the emergence and evolution of many viruses, which reinforces the fundamental need to investigate the virome of these animals. It appears reasonable to assume that the diversity of the viruses of the genus Hepatovirus has yet to be fully understood12. Records from the DBatVir platform indicate that 321 picornavirus sequences have been deposited in this database, of which, 48 refer to the genus Hepatovirus, although the bat genus Artibeus was not cited in any of these records13.
The present study provides the description and identification of the genome of a new hepatovirus, denominated Hepatovirus H isolate sotense, retrieved from a specimen of the flat-faced fruit-eating bat, Artibeus planirostris, collected in the Maranhão State, Mid-North region of Brazil.
Results
Sampling
The material analyzed here consisted of 16 samples (pools) of internal organs (liver and lungs) obtained from four bat species of the genus Artibeus from different localities in the Mid-North region of Brazil—Artibeus obscurus (Pool 2—Timon and Pool 8—Carolina), Artibeus cinereus (Pool 1—Timon and Pool 3—Caxias), Artibeus lituratus (Pool 4—Gurupi, Pool 6—Timon, and Pool 10—Carolina), and A. planirostris (Pool 5—São João do Sóter, Pool 7—Timon, Pool 9, 11–16—Carolina). For the sample of the species A. planirostris (Pool 5—São João do Sóter), the genome of a new hepatovirus, called Hepatovirus H isolate sotense, was described and identified.
Sequencing and bioinformatic analyses
The genomic libraries (cDNA) were constructed based on a total of 54,681,026 million read, on average, with these reads being used to compile a de novo arrangement. The taxonomic classification programs estimated 54,68,926 microbial reads, with the lowest number—45,919—being recorded for viruses. When visualized in the MEGAN software, the alignment indicated considerable viral diversity, with reads corresponding to 39 partial sequences related to RNA viruses, belonging principally to the families Retroviridae, Mimiviridae, Siphoviridae and Myoviridae, as shown in the heat map (Fig. 1). The distribution of these families in the heat map indicates the presence of the complete Hepatovirus genome in pool 5, which will be analyzed and described in detail below.

Heat map showing the distribution of the reads for the different viral families. The figure shows the viral families that were detected. The number of reads is indicated by the color scale, which ranges from light yellow, for the families with the smallest number of reads, to bright red, which indicates the largest number of reads. The Retroviridae, Mimiviridae, Siphoviridae, and Myoviridae were the most frequent viral families.
High-throughput sequencing
The genome of 7390 nucleotides (nt) has untranslated regions (UTRs) of 737 nt and 64 nt at the 5' and 3' ends, respectively. The mean genome coverage was 26×, with a GC content of 36.6%. The analysis of the open reading frame matrix identified a protein with 2000 predicted amino acids and a molecular weight of 248,690 kDa. This genome has been deposited in the GenBank public database under the name Hepatovirus H isolate sotense (Accession number: OR367421), with the codes BioProject PRJNA947063, BioSample: SAMN36696548, Sequence Read Archive: SRR24657753. The raw metagenomic data are available on the code OR367421 for Hepatovirus H isolate sotense, in the link (https://www.ncbi.nlm.nih.gov/nuccore/OR367421). The BlastX result indicated an identity of 76.8% with Hepatovirus H2 (Accession number: KT452714), isolated from a bat (Eidolon helvum) in Ghana in 2010.
Phylogenetic and similarity analysis
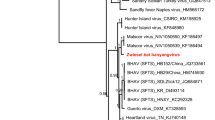
The phylogenetic analyses revealed two major clades formed by Hepatovirus A, F, D, I and Hepatovirus H, G, B, E, C (Fig. 2). Hepatovirus H isolate sotense was assigned to a monophyletic clade within the Hepatovirus H species, however it occupied the most basal branch of the clade, with high (100%) bootstrap support.

Phylogenetic neighbor-joining tree of the Hepatovirus species based on their amino acid sequences, compiled in IG-TREE com 1000 bootstrap replicates. The sequence obtained in the present study is highlighted in italics, with two asterisks.
A matrix was compiled which grouped the hepatoviruses that are closest to H. H isolate sotense, which showed that this species was most similar to the sequence of Hepatovirus H2 (72.7% nt and 76.8% aa) and Bat Hepatovirus G (64.9% nt and 66.5% aa). The values found here indicate clearly that, although H. H isolate sotense is closest to hepatoviruses H2 but, it is clearly different from it, which reinforces its status as a new genotype of Hepatovirus H species (Table 1).
Given the overall pattern found in the phylogenetic tree and this matrix, a new analysis of similarity was run to verify the level of differentiation among the Hepatovirus species, correlating the amino acid and nucleotide sequences of the ORF. This analysis indicated that the sequence of Hepatovirus H isolate sotense is closely related to those of the other viruses of genus, but is also clearly distinct from them. The identity (differences per site between the sequences) of the species ranged from 55.5 to 99.7% in the case of the amino acids and 50.0–99.9% for the nucleotides. Considering only the clade that unites the H species in the phylogenetic tree, the identity of the amino acids was 71.3–98.0%, while that of the nucleotides was 74.9–99.6% (Supplementary Table 1). When only the P1 region of the polyprotein was considered in this comparison, the values were 60.5–99.9% and 64.0–99.9%, respectively. In the specific case of Hepatovirus H, the ranges were 85.0–99.6% and 75.0–96.8%, respectively (Supplementary Table 2). One last matrix was compiled, considering the 2C+3CD regions of the polyprotein, and once again, the same pattern was found, with an identity of 52.0–99.8% for the nucleotides and 57.0–99.6% for the amino acids, considering all the heptoviruses, and 77.6–99.5% and 71.5–98.1%, respectively, for Hepatovirus H (Supplementary Table 3).
Genomic characterization
The ordination of the sequences revealed the classic organization of the genome of the members of the genus Hepatovirus, which confirms the classification of Hepatovirus H isolate sotense as a picornavirus. The structure identified in this analysis indicated the presence of a single ORF, which codifies a polyprotein. The viral polyprotein is divided into three regions: P1, P2, and P3. The P1 region encodes the viral capsid proteins (VP4-VP2-VP3-VP1), whilst the P2 and P3 regions encode proteins involved in protein processing (2Apro, 3Cpro and 3CDpro) and genome replication (2B, 2C, 3AB, 3BVPg, 3CDpro, 3Dpol)14. The proteolytic cleavage map was derived from the alignment by analogy with the other hepatovirus sequences that grouped with Hepatovirus H isolate sotense in the analysis presented here. While the organization of the genome of Hepatovirus H isolate sotense is typical of that of other hepatoviruses, it was differentiated from the other members of the genus Hepatovirus. In particular, there is a high level of variation in the specific protein-codifying regions of the different species of the genus. As can be seen here, Hepatovirus H isolate sotense has a poorly-conserved region downstream from VP1, in comparison with the other hepatoviruses analyzed here (Supplementary Fig. 2), which is symptomatic of the differentiation of the species of this genus. The mean level of similarity of the proteins found in the different hepatovirus species, including Hepatovirus H isolate sotense, is shown in Table 1 (see also Fig. 2). The analysis of the 10 sequences (Hepatovirus H isolate sotense, Phopivirus; Hedgehog hepatovirus Igel8Erieur2014; Hedgehog hepatovirus; Hepatovirus H2; Hepatovirus G2; Bat hepatovirus BUO2BF86Colafr2010; Rodent hepatovirus CIV459Lopsik2004; Bat hepatovirus SMG18520Minmav2014; Tupaia hepatovirus A) showed that they were, in general, less similar to one another than those of the five sequences (Hepatovirus H isolate sotense; Hedgehog hepatovirus; Hedgehog hepatovirus Igel8Erieur2014; Hepatovirus H2; Tupaia hepatovirus A). This indicates that the species of the latter group are more closely-related, even though they are clearly differentiated.
The nucleotide sequences of the four most closely related Hepatovirus genotypes in the phylogenetic tree (Hedgehog hepatovirus, KT452691; Hedgehog hepatovirus Igel8Erieur2014, KT452698; Hepatovirus H2, KT452714; and Tupaia hepatovirus A, KT877158) were analyzed using the Simplot software, with Hepatovirus H isolate sotense serving as the reference sequence. The similarity plot resulting from this analysis (Fig. 3) indicates that a substantial similarity, with mean percentages in the upper 70%, in the VP1 region of Hepatovirus H isolate sotense was observed within this species. Subsequently, the P3 region exhibited relatively lower similarity, while the lowest similarity, around 70%, was observed in the P2 region.

Similarity analysis using SimPlot. The figure illustrates the SimPlot analysis of the query sequence of Hepatovirus H isolate sotense genotype CH-APSJS/2021 (NCBI ID OR367421). Identification of all proteins is presented at the top of the chart. The analysis employed the following parameters: Window: 200 bp, Step: 20 bp, GapStrip: On, Kimura (2-parameter), T/t: 2.0. The alignment positions indicated in nucleotides (nt).
The analysis of the functional domains indicated that Hepatovirus H isolate sotense shares its pattern of genomic organization with the samples that were most closely related in the phylogenetic analysis. This further confirms the taxonomic status of this species within the genus Hepatovirus (Fig. 4).

Demonstration of the different functional domains of the polyprotein from various viruses belonging to the Hepatovirus species. The yellow arrow corresponds to the coding region of the polyprotein. The different domains are depicted in colors according to the identified protein, and their respective protein codes are in parentheses.
The results of the present study are part of a larger project that is describing the viromes of different species of bat. In general, the isolation of hepatoviruses, and other hepatotrophic viruses, is a major challenge, even considering the most common cell lines. In the specific case of bats, few studies have been able to isolate effectively the hepatoviruses of these animals, although, in the future, when the techniques have been fully standardized, the target viruses will be isolated from the FRhK-4 cells of all the samples in which some type of viral infection has been confirmed by immunofluorescence.
In the specific case of the Artibeus species analyzed here, samples of the lung, liver, heart, spleen, intestine, brain, kidney, and blood were examined by RT-PCR to determine the presence of viruses in specific organs.
Discussion
The results of the present study indicate the discovery of a novel isolate of Hepatovirus H, in the genus Hepatovirus. These hepatoviruses are known to include many pathogens of both humans and other vertebrates, causing diseases that affect mainly the liver15. While many new Hepatovirus species have been found recently using new generation sequencing, the pathogenicity of most of these viruses is still relatively poorly understood16. As chiropterans are known to act as reservoirs of an ample variety of viral agents5, it will be extremely important to identify and describe the viromes of the different species of this mammalian order, to provide an ever more solid foundation for further research in epidemiology and public health. As far as we know, this is the first report of a Hepatovirus in bats from South America, considering that the only previous description was from Central America (Costa Rica), with the virus occurring in Natalus lanatus, with sequences being obtained from partial samples of two isolates, CR10401 (KT452716.1) and CR10366 (KT452715.1)16. Furthermore, according to data from the DBatVir Platform researched in November 202313, there are no records of Hepatovirus in any of the species of the genus Artibeus, with the first being described here.
A number of new Hepatovirus species have been described in recent years, resulting in an exponential growth in the known diversity of picornaviruses, as well as an ample variety of hosts, distributed throughout the world, that can be parasitized by these viruses16. One study of small mammals of three different orders16 identified 13 new Hepatovirus species, including four that infect bats. Two other new species of Hepatovirus have been identified from specimens of seals17, and one new species was described in a study of marmots18. In the specific case of the Bat Hepatovirus, a recent study19 identified a new species in Hipposideros armiger from China, while a second study20 recorded a new species in Eptesicus fuscus in the United States. It is worth noting that of all these species, Hepatovirus H isolate sotense obtained in the present study was shown to be most closely related to Hepatovirus G. Based on the data from the International Committee on Taxonomy of Viruses (ICTV), only Hepatovirus A is capable of causing natural infectious hepatitis in humans, and experimental infection in other primatas, although persistent infection does not occur in vivo, and the viruses are not associated with chronic hepatitis. Few biological data are available on the other hepatoviruses, however, or the clinical manifestations they cause in their hosts14. While there has been a considerable recent increase in the number of species, these new hepatoviruses have been assigned clearly to the respective genus, given the greater distances that separate them from the genus Tremovirus, which represents the closest picornavirid lineage, as well as the criteria adopted by the ICTV for the classification of picornaviruses9.
It is important to note here that, for the ICTV to accept the proposal of these new species, for the classification of the new hepatovirus variants, it was necessary to provide complete genome sequences, and demonstrate their clear separation in the phylogenetic analyses21. In the present study, it was demonstrated clearly that the proposed taxon is a new genotype belonging to Hepatovirus H, here named Hepatovirus H isolate sotense, taking the findings of the present study into consideration. These results showed that the isolate was less than 30% divergent in the polyprotein amino acid sequence, less than 30% divergent in the P1 amino acid sequence, less than 30% divergent in the 2C + 3CD amino acid sequence, and shared a common genomic organization with the other species analyzed. These criteria were adopted to define the members of a species in the genus Hepatovirus.
The complexity of the genealogy of the hepatoviruses may be at least partly a result of the frequent changes in host that occurred in the distant past of the evolutionary history of the viruses22. The phylogeny presented here confirms this pattern, given that the viruses tended to group according to their different hosts, as shown clearly by the fact that the most basal clade is composed of the viruses of rodent, bat (including A. planirostris), hedgehog, tree shrew, and seal hosts, with high levels of statistical support. This analysis clearly demonstrates that Hepatovirus H isolate sotense belongs to the genus Hepatovirus, more specifically to species H, given that the statistical values support this claim. However, it is a new strain, as it did not form a monophyletic cluster with the other representatives of the species.
The only clearly defined clade in the phylogenetic tree is that of the human Hepatovirus A (HAV). Recent studies indicate that the evolutionary history of the HAVs is distinct from that of the other hepatoviruses described up to now, with differences in the nucleotide sequence of the genome and the structure of the capsid, characteristics that are shared with a primitive insect virus23. These viruses are also capable of releasing infected cells that appear to be enclosed in a membrane, to become virions, which are almost completely enveloped, a unique trait among the picornaviruses. In addition, cells infected by HAV may release vesicles coated with a membrane that contains a virion, resulting in unusual virions enveloped in a membrane, which are not observed in other picornaviruses24.
Overall, then, the sum of the evidence presented here supports the recognition of a new Hepatovirus H virus, denominated Hepatovirus H isolate sotense, which was isolated from the internal organ sample (liver and lung) of a specimen of the fruit-eating bat, Artibeus planirostris, and was clearly a picornavirus with genomic structure typical of the hepatoviruses. This is an important discovery, not least because this is the first study of its kind from the Brazilian state of Maranhão, but also because most previous studies of viruses associated with bats in this state have focused on species of the family Rhabdoviridae. It is important to note here that all the specimens collected for the present study tested negative for the rabies virus. It is important to note here that previous studies22 have shown that hepatoviruses are capable of swapping hosts. In this case, the species described here deserves further consideration, given that among the species of Hepatovirus that infect bats (C, E, G, and H), species H is the one that has the ability to infect different vertebrate species, in addition to bats, and is therefore more prone to the occurrence of mutations in its genome, and thus demands attention from the state’s epidemiological monitoring agencies. It is also important to note, in this context, that the specimen analyzed here did not present any clinical manifestations of the disease, such as an inflamed liver, or other signs of ongoing infection, which is typical of many viral pathogens in bats, and hampers the diagnosis of infection based solely on the behavior of the individual.
Conclusions
While the number of studies of viral diversity in Brazil has increased steadily in recent decades, and in particular following the COVID-19 pandemic, research in this field is still incipient, especially in the state of Maranhão. Studies of the viruses of wild animals are important, not only to catalog this diversity, but also to identify species (as in the case of the hepatoviruses) that are potentially pathogenic for animals and humans. This knowledge will be fundamental for the elaboration of effective public health strategies, and the development of measures for the prevention or eventual mitigation of outbreaks, given that epidemiological studies are still not focused on the origins of the disease, that is, they tend to focus on outbreaks, rather than prevention. This is worrying, from an epidemiological perspective, and means that the effective prevention and control of diseases will depend on a more systematic approach, linking the health of humans, animals, and the environment. Overall, the findings of the present study provide important new insights into the diversity, hosts, and geographic distribution of the genus Hepatovirus, as the range of occurrence of this virus in bats increases to South America.
Materials and methods
Ethics statement
Field procedures and protocols for trapping, euthanasia and tissue processing, conforming to the guidelines of the National Council for the Control of Animal Experimentation (CONCEA), aiming to comply with the animal welfare standards of CFMV Resolution No. 1000/2012 and Law 11,794/2006. The collection of specimens was authorized by federal license ICMBIO/SISBIO No. 68047-2. The animals were sedated with a combination of ketamine and xylazine intramuscularly and subsequently euthanized with an overdose of lidocaine in the foramen magnum. The collection of the animal viscera (a pool of lung and liver) was carried out by veterinarians, in compliance with biosafety standards. The samples collected were stored in cryogenic vials, which were duly identified and stored in liquid nitrogen for later forwarding to the Arbovirology and Hemorrhagic Fevers laboratory of the Instituto Evandro Chagas (IEC), where the samples were handled in a laminar flow cabinet at biosafety level 2 by personnel trained in the use of personal protective equipment, with viscera maceration taking place in the NB3 laboratory. The Ethics Committee Council of CEMAVE and SISBIO reviewed and approved the field protocols and experimental procedures through authorization protocol 025/2019.
Sample collection area
The material analyzed here consisted of 16 samples (pools) of viscera (liver and lungs) obtained from four bat species of the genus Artibeus—A. obscurus (Pool 2—Timon and Pool 8—Carolina), A. cinereus (Pool 1—Timon and Pool 3—Caxias), A. lituratus (Pool 4—Gurupi, Pool 6—Timon, and Pool 10—Carolina), and A. planirostris (Pool 5—São João do Sóter, Pool 7—Timon, Pool 9, 11–16—Carolina). Each pool contained up to five tissue samples, depending on the species and collecting locality. The specimen of A. planirostris analyzed in the present study was collected in a mist net on 28-Oct-2021 in the municipality of São João do Sóter (Fig. 5), during the capture of specimens in areas of the Amazon and Cerrado biomes in the northern Brazilian state of Maranhão, as part of a project investigating viral pathogens in bats of the genus Artibeus. The sample from which the complete sequence of Hepatovirus H isolate sotense was obtained from pool 5 corresponds to a young adult male A. planirostris, with a body weight of 29 g and no apparent symptoms of any zoonosis known to affect bats, such as excessive salivation, lethargic or aggressive behavior, and alterations of the size or coloration of the internal organs.

Artibeus planirostris collecting locality in the Brazilian state of Maranhão. Map produced in QGIS 3.22.4
Metagenomic sequencing
For analysis, the genetic material was extracted from combined samples of liver and lung tissue, which were macerated together with 1 mL of the TRIzol™ Plus RNA Purification kit and a 5-mm diameter steel sphere by agitation in a TissueLyser II. The extracted RNA was purified and isolated using a PureLink RNA Mini kit, following the manufacturer’s instructions. The simple and double-strand cDNA was synthesized using the SuperScriptTM VILOTM Master Mix (Thermo Fischer Scientific, Waltham, MA, USA) and NEBNext® mRNA Second Strand Synthesis Module (New England BioLabs, Ipswich, MA, USA) kits, respectively.
Genomic data analysis
The genomic library was prepared following the guidelines of the Nextera XT DNA kit, and sequenced in the NextSeq 500 platform (Illumina, Inc) using the NextSeq 500/550 High Output kit v2.5 (300 cycles) and the paired-end approach, following the manufacturer’s recommendations.
The quality of the raw data was evaluated by first using Fastp v0.23.225 to remove the short reads (less than 50 nt), fragments of adaptors and reads with indeterminate bases (reads with more than 10% of the N).
The ribosomal RNA was then removed in SortMeRNA v2.126. The processed reads were then compared with the non-redundant (nr) amino acid sequence database of the NCBI using DIAMOND v2.1.727 with an e-value of 1e−4. The DIAMOND output file was visualized in MEGAN628.
The reads were also mounted using the de novo approach in SPAdes v3.13.129 and Megahit v1.2.930, with k-mer values of 21, 33, 55 and 77 and of 21, 31, 41, 51, 61, 71, 81, 91, and 99. The grouped contigs (scaffolds) were mapped in DIAMOND27 with the same parameters used for the reads, and then inspected in MEGAN617. The contigs that were similar to related viral sequences were evaluated in Geneious v9.1.831.
Phylogenetic analyses
The phylogenetic inference was based on the amino acid sequences of the different Hepatovirus strains available in the NCBI database, using the protein-codifying region. The datasets generated here were submitted, together with the present sample, to multiple sequence alignment (MSA) in Mafft v.732. The result of this alignment was inspected manually, and corrected when necessary, in Geneious v9.1.831.
The aligned dataset was initially analyzed to identify the best nucleotide substitution model before constructing the phylogenetic trees using the Maximum Likelihood (ML) method33. Both these analyses were run in IQ-TREE v1.6.1234.
The ML analysis was complemented with a bootstrap test, based on 1000 replicates, to determine the reliability of the groups identified in the analysis35. The phylogeny was visualized in FigTree v1.4.436. It was decided not to use a root sequence for the dataset, but rather a mid-point rooting approach, which is available in the phylogeny visualization program. Following the evaluation and edition of the phylogeny, an “.svg” (Scalable Vector Graphics) file was generated for the edition and adjustment of the image in Inkscape v1.237. All procedures were were conducted in accordance with the relevant guidelines and regulations.
Data availability
GenBank accession numbers. The raw metagenomic data are available on the code OR367421 for Hepatovirus H isolate sotense, BioProject PRJNA947063, BioSample: SAMN36696548, Sequence Read Archive: SRR24657753 in the link https://www.ncbi.nlm.nih.gov/nuccore/OR367421. The data are simultaneously made available to other INSDC databases, the European Nucleotide Archive (ENA) and the DNA Data Bank of Japan (DDBJ).
References
Taylor, M., Tuttle, M. D. BATS: An Illustrated Guide to All Species. 4rd ed. 403 (Science Editor & Photographer, 2019).
Reis, N. R., Peracchi, A. L., Batista, C. B., Lima, I. P. & Pereira, A. D. História Natural dos Morcegos Brasileiros Chave de Identificação de Espécies (Technical Books Editora, Brasil, 2017).
Jorge, T. M. R., da Silva, A. V. C. & Ortêncio Filho, H. Interação morcego-planta: Uma análise cienciométrica de estudos no Brasil. ICSA 6(1), 43–52 (2017).
Wang, L. F. & Anderson, D. E. Viruses in bats and potential spillover to animals and humans. Curr. Opin. Virol. 34, 79–89 (2019).
Mollentze, N. & Streicker, D. G. Viral zoonotic risk is homogenous among taxonomic orders of mammalian and avian reservoir hosts. PNAS 28(17), 9423–9430. https://doi.org/10.1073/pnas.1919176117 (2020).
Abreu, W. U. et al. Sequências retrovirais de interesse em medicina veterinária identificadas em morcegos urbanos da Amazônia. Res. Soc. Dev. 11, 10 (2022).
Koonin, E. V. et al. Global organization and proposed megataxonomy of the virus world. Microbiol. Mol. Biol. Rev. 84, e00061-e119 (2020).
Resources: Picornavírus. https://www.picornaviridae.com//index.html.
Zell, R. et al. ICTV virus taxonomy profile: Picornaviridae. J. Gen. Virol. 98, 2421–2422 (2017).
Wang, Y. et al. The fecal virome of red-crowned cranes. Arch. Virol. 164, 3–16. https://doi.org/10.1007/s00705-018-4037-x (2019).
Shi, M. et al. Redefining the invertebrate RNA virosphere. Nature. 540, 539–543 (2016).
Yinda, C. K. et al. Highly diverse population of Picornaviridae and other members of the Picornavirales, in Cameroonian fruit bats. BMC Genom. 18, 249 (2017).
Resources: Picornavírus. http://www.mgc.ac.cn/cgi-bin/DBatVir/main.cgi. Accessed 16 July 2023 (2023).
International Committee on Taxonomy of Viruses (ICTV). https://ictv.global/report/chapter/picornaviridae/picornaviridae/hepatovirus. Accessed 14 July 2023 (2023).
Zell, R. Picornaviridae—The ever-growing virus family. Arch. Virol. 163, 299–317 (2018).
Zhao, S. et al. Senecavirus A enhances its adaptive evolution via synonymous codon bias evolution. Viruses 14(5), 1055. https://doi.org/10.3390/v14051055 (2022).
Drexler, J. F. et al. Evolutionary origins of hepatitis A virus in small mammals. Proc. Natl. Acad. Sci. USA 112(49), 15190–15195. https://doi.org/10.1073/pnas.1516992112 (2015).
Anthony, S. J. et al. Discovery of a novel Hepatovirus (phopivirus of seals) related to human hepatitis A virus. mBio 6, 4. https://doi.org/10.1128/mBio.01180-15 (2015).
Yu, J. M. et al. A novel hepatovirus identified in wild woodchuck Marmota himalayana. Sci. Rep. 6, 22361. https://doi.org/10.1038/srep22361 (2016).
Li, W. et al. Genomic characterization of a novel Hepatovirus from great roundleaf bats in China. Virol. Sin. 33(1), 108–110. https://doi.org/10.1007/s12250-018-0013-6 (2018).
Temeeyasen, G. & Hause, B. M. Novel hepatoviruses in synanthropic bats in the upper Midwestern United States. Arch. Virol. 167(12), 2749–2751. https://doi.org/10.1007/s00705-022-05610-8 (2022).
Smith, D. B. & Simmonds, P. Classification and genomic diversity of enterically transmitted hepatitis viruses. Cold Spring Harb. Perspect. Med. 4, 8. https://doi.org/10.1101/cshperspect.a031880 (2018).
Sander, A. L., Corman, V. M., Lukashev, A. N. & Drexler, J. F. Evolutionary origins of enteric hepatitis viruses. Cold Spring Harb. Perspect. Med. https://doi.org/10.1101/cshperspect.a031690 (2018).
Stuart, D. I., Ren, J., Wang, X., Rao, Z. & Fry, E. E. Hepatitis A virus capsid structure. Cold Spring Harb. Perspect. Med. https://doi.org/10.1101/cshperspect.a031807 (2018).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. Fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34(17), i884–i890 (2018).
Kopylova, E., Noé, L. & Touzet, H. SortMeRNA: Fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics 28(24), 3211–3217. https://doi.org/10.1093/bioinformatics/bts611 (2012).
Buchfink, B., Xie, C. & Huson, D. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60. https://doi.org/10.1038/nmeth.3176 (2015).
Huson, D. H., Auch, A. F., Qi, J. & Schuster, S. C. MEGAN analysis of metagenomic data. Genome Res. 17(3), 377–386. https://doi.org/10.1101/gr.5969107 (2007).
Bankevich, A. et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 5 (2012).
Li, D. et al. MEGAHIT v.10: A fast and scalable metagenome assembler driven by advanced methodologies and community practices. Methods. 102, 3–11 (2016).
Geneious. https://www.geneious.com/. Accessed 14 July 2023 (2023).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013).
Myung, I. J. Tutorial on maximum likelihood estimation. J. Math. Psychol. 47, 90–100 (2003).
Nguyen, L. T., Schmidt, H. A., Haeseler, A. & Minh, B. Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum likelihood phylogenies. Mol. Biol. Evol. 32, 268–274 (2015).
Felsenstein, J. Confidence limits on phylogenies: An approach using the bootstrap. Evolution 39, 783–791 (1985).
FigTree v1.4.4. https://github.com/rambaut/figtree/releases/tag/v1.4.4. Accessed 14 July 2023 (2023).
Inkscape v.1.1. https://inkscape.org/release/inkscape-1.1. Accessed 14 July 2023 (2023).
Acknowledgements
The authors are grateful to the “Bats do Maranhão” team, formed by Samira Brito, Cleison Costa, Fábio Cardoso, Priscila Olímpio, Amanda Cristiny, Samuel Coutinho, Marcelo Ventura, Walna Pires, Marxo Morais and Aglay Morgana, who helped collect the specimens.
Funding
The manuscript was funded by PROPESP/UFPA (PAPQ), Fundação de Amparo à Pesquisa e ao Desenvolvimento Científico e Tecnológico do Maranhão (Grant Nos. 02724/20, 002/2019), Programa Nacional de Cooperação Acadêmica na Amazônia (23038.005350/2018-78), Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (88887.761304/2022-00), Conselho Nacional de Desenvolvimento Científico e Tecnológico (314522/2021-2) and Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) (309916/2021-6).
Author information
Authors and Affiliations
Contributions
Conceptualization, Walna Micaelle de Moraes Pires, Ana Cecília Ribeiro Cruz, Alex Junior Souza Souza, Sandro Silva, Claudene Barros; methods, Walna Micaelle de Moraes Pires, Ana Cecília Ribeiro Cruz, Sandro Silva, Samira Mendes; Taciana Fernandes Souza Barbosa Coelho; software, Walna Micaelle de Moraes Pires, Sandro Silva; Walna Micaelle de Moraes Pires, Sandro Silva; formal analyses, Walna Micaelle de Moraes Pires, Sandro Silva and Daniel Dias; investigation, Walna Micaelle de Moraes Pires, Sandro Silva, Daniel Dias and José Wilson Rosa Júnior; resources, Iracilda Sampaio, Ana Cecília Ribeiro Cruz, Elmary da Costa Fraga, Claudene Barros; data curation, Walna Micaelle de Moraes Pires, Sandro Silva, Daniel Dias and José Wilson Rosa Júnior; redaction—preparation of the original draft, Walna Micaelle de Moraes Pires; redaction—revision and edition, Walna Micaelle de Moraes Pires, Ana Cecília Ribeiro Cruz, Alex Junior Souza Souza, Sandro Silva, Claudene Barros, Iracilda Sampaio; visualization, Ana Cecília Ribeiro Cruz, Claudene Barros, Iracilda Sampaio.; supervision, Ana Cecília Ribeiro Cruz and Sandro Silva; administration of the projects, Claudene Barros, Elmary da Costa Fraga, Iracilda Sampaio; funding acquisition,Ana Cecília Ribeiro Cruz, Claudene Barros, Elmary da Costa Fraga, Iracilda Sampaio. All the authors have read and agreed to the publication of this version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
de Moraes Pires, W.M., Cruz, A.C.R., de Souza, A.J.S. et al. Genomic characterization of a novel Hepatovirus identified in Maranhão state, Brazil. Sci Rep 14, 7981 (2024). https://doi.org/10.1038/s41598-024-58171-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-58171-y
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
