
Abstract
Dual-specificity tyrosine phosphorylation-regulated kinase 1 A (DYRK1A) is essential for human development, and DYRK1A haploinsufficiency is associated with a recognizable developmental syndrome and variable clinical features. Here, we present a patient with DYRK1A haploinsufficiency syndrome, including facial dysmorphism, delayed motor development, cardiovascular system defects, and brain atrophy. Exome sequencing identified a novel de novo heterozygous mutation of the human DYRK1A gene (c.1185dup), which generated a translational termination codon and resulted in a C-terminally truncated protein (DYRK1A-E396ter). To study the molecular effect of this truncation, we generated mammalian cell and Drosophila models that recapitulated the DYRK1A protein truncation. Analysis of the structure and deformation energy of the mutant protein predicted a reduction in protein stability. Experimentally, the mutant protein was efficiently degraded by the ubiquitin-dependent proteasome pathway and was barely detectable in mammalian cells. More importantly, the mutant kinase was intrinsically inactive and had little negative impact on the wild-type protein. Similarly, the mutant protein had a minimal effect on Drosophila phenotypes, confirming its loss-of-function in vivo. Together, our results suggest that the novel heterozygous mutation of DYRK1A resulted in loss-of-function of the kinase activity of DYRK1A and may contribute to the developmental delay observed in the patient.
Similar content being viewed by others

Introduction
Dual-specificity tyrosine phosphorylation-regulated kinase 1A (DYRK1A) is a member of the highly conserved DYRK kinase family that belongs to the CMGC kinase superfamily. DYRK1A phosphorylates serine and threonine residues on its target substrates and autophosphorylates its own tyrosine residues1,2. Due to the variety of proteins that DYRK1A phosphorylates, it plays essential roles in a wide range of cellular signalling pathways and processes, including neurogenesis, neuronal differentiation and proliferation3, synaptic transmission4, cell cycle5, apoptosis6, splicing7, and RNA transcription8. Many of these pathways and processes regulate neurological function, and, indeed, DYRK1A overexpression or overactivity is associated with many neurodegenerative diseases, such as Alzheimer’s, Parkinson’s, Huntington’s, and Pick’s diseases, and even certain types of cancers (e.g., glioma)9,10,11,12,13,14. DYRK1A is also thought to contribute to several of the phenotypic and neurocognitive features of Down Syndrome (DS), because individuals with DS carry an extra copy of DYRK1A15,16,17. Decreased DYRK1A expression or activity is also pathogenic. For example, mice hemizygous for DYRK1A have substantial phenotypic defects, including smaller body size, microcephaly, reduced numbers of neurons, abnormal motor function, gait disturbances, and impaired cognitive function18,19. Human DYRK1A haploinsufficiency is generated by a variety of mutations and is a potential cause of a recognizable developmental syndrome that is characterized by variable clinical features, including intellectual disability, developmental delay, microcephaly, dysmorphic facial features, speech delay, autism, febrile seizures, and ocular malformations (OMIM: 614104, ORPHANET: 464306)20,21. Individuals with this syndrome were first identified with partial monosomies of chromosome 21 on routine karyotypes that encompassed the DYRK1A gene (21q22.13)22. More recently, the diagnosis of numerous de novo mutations in DYRK1A has been achieved by next generation sequencing, which has facilitated and broadened the clinical characterization of DYRK1A disruptions. To date, many mutations associated with DYRK1A have been identified and include gross deletions, small deletions, point mutations, complex rearrangements, small indels, and splice-site mutations (Human Gene Mutation Database, http://www.hgmd.org). Many of these mutations result in truncated proteins that partially or completely lack the DYRK1A kinase domain and thereby lose their catalytic activity.
Here, we report a novel de novo DYRK1A mutation occurring in the β-sheet of the CMGC insert, which is located in the C-terminal end of the kinase domain. This nonsense mutation led to the production of a C-terminally truncated kinase domain protein (DYRK1A-E396ter). The resulting mutant protein was not only efficiently degraded by the proteasome but was also catalytically inactive in mammalian cell and fly models, indicating complete loss-of-function of DYRK1A.
Materials and Methods
Patient
The study was approved by the Institutional Review Board of Pusan National University Yangsan Hospital (approval number: 05-2019-103) and adhered to the tenets of the Declaration of Helsinki involving ethical principles for medical research with human subjects. Informed consent was obtained from the child’s parents.
Genetic analysis
Written informed consent was obtained from all participants before blood was drawn. Genomic DNA was isolated using the QIAamp DNA Blood Midi kit (Qiagen, Hilden, Germany) from participants’ leukocytes in the peripheral blood, according to the manufacturer’s standard protocols. The extracted gDNA was evaluated using the TruSight One Sequencing Panel (Illumina Inc., San Diego, CA, USA) as described previously23. Captured targeted regions were sequenced using the Hiseq 2500 Sequencing System (Illumina Inc.) following the manufacturer’s instructions. Alignment and variant calling was done automatically by on-instrument tools. Imported sequence data was filtered for specified genes and converted into a customized report using the VariantStudio software. Pathogenic variants were evaluated by the practical statement released by the American College of Medical Genetics and Genomics24.
Plasmid construction
To construct plasmids expressing FLAG-DYRK1A proteins, the DNA fragment encoding FLAG (DYKDDDDK) was inserted into a pcDNA3.1(+) vector at NheI/HindIII sites, and the open reading frame of human DYRK1A (NM_001396.4) was cloned into a pcDNA3.1(+) vector at HindIII/XbaI sites. Plasmids expressing FLAG-DYRK1A-E396ter and FLAG-DYRK1A-K188R were generated by mutating the original sequence with a QuikChange II Site-Directed Mutagenesis Kit (Agilent Technologies, Santa Clara, CA, USA), according to the manufacturer’s method. The following primers that are specific to each mutant were used: 5ʹ-CAAAAGCAAGAAAGTTCTTTTGAGAAGTTGCCAGATG-3’ (forward) and 5ʹ-CATCTGGCAACTTCTCAAAAGAACTTTCTTGCTTTTG-3ʹ (reverse) for FLAG-DYRK1A-E396ter; 5ʹ-CAAGAATGGGTTGCCATTAGAATAATAAAGAACAAGAAG-3ʹ (forward) and 5ʹ-CTTCTTGTTCTTTATTATTCTAATGGCAACCCATTCTTG-3ʹ (reverse) for FLAG-DYRK1A-K188R.
Cell culture and transfection
Human embryonic kidney 293T cells were cultured in Dulbecco’s Modified Eagle’s Medium containing 10% foetal bovine serum (Welgene, Gyeongsan-si, Gyeongsangbuk-do, Republic of Korea) supplemented with 1% streptomycin and penicillin. The cells were seeded at approximately 50% confluency into cell culture plates and were maintained overnight at 37 °C under 5% CO2. When the cells reached 60–80% confluency, they were transfected with plasmids using the XtremeGene Transfection Reagent (Roche, Basel, Switzerland), according to the manufacturer’s instructions. Transfected cells were incubated at 37 °C for 24 h prior to harvest or analysis.
Chemicals
We used the proteasome inhibitor MG132 (Calbiochem, San Diego, CA, USA), the lysosomal inhibitor NH4Cl (Sigma-Aldrich, St. Louis, MO, USA), the calpain inhibitor calpeptin (Calbiochem), and the autophagy inhibitor 3-methyladenine (Sigma-Aldrich) for protein degradation pathway analyses. All chemicals were dissolved in dimethyl sulfoxide (DMSO) prior to treatment, and the cells were treated with 10 μM of each compound for 15 h after plasmid transfection.
Quantitative western blot analysis
Quantitative western blot analysis was performed as described previously25. The following antibodies were used: anti-FLAG M2 antibody (catalogue #F1804; Sigma-Aldrich; 1:500 dilution), anti-HA antibody (catalogue #sc-805; Santa Cruz Biotechnology, Santa Cruz, CA, USA; 1:1,000 dilution), anti-hnRNP A1 antibody (catalogue #CSB-PA002942; Cusabio, Houston, TX, USA; 1:1,000 dilution), anti-Tau antibody (catalogue #MN1040; Thermo Fisher Scientific, Waltham, MA, USA; 1:1,000 dilution), and anti-phospho-Tau (at residue Thr-212) antibody (catalogue # 4–740 G; Thermo Fisher Scientific; 1:1,000 dilution). Signals were detected on a LAS-4000 image analyser using a ClarityTM Western ECL Substrate (BIO-RAD, Hercules, CA, USA) and then quantitatively analysed using the NIH ImageJ software (National Institutes of Health, Bethesda, MD, USA).
Quantitative RT-PCR (qRT-PCR)
The 293T cells were seeded in 6-well cell culture plates and transfected with FLAG-tagged wild-type or mutant DYRK1A plasmids for 24 h. TRIzol reagent (Invitrogen, Carlsbad, CA, USA) was used for total RNA extraction, and the Omniscript RT Kit (Qiagen, Hilden, Germany) was used to synthesize first-strand cDNA from 1 μg of total RNA, which was primed with oligo-dT primers. qRT-PCR was performed using the TOPreal SYBR Green PCR Kit (Enzynomics, Daejeon, Republic of Korea) in triplicate with the following primer sets: 5ʹ-TAGCATGGATTACAAGGATGACGATG-3ʹ (FLAG-DYRK1A forward), 5ʹ- ACATAAGTGACCAACAGGTTTCTGC-3ʹ (FLAG-DYRK1A reverse), 5ʹ-AGAGCTACGAGCTGCCTGAC-3ʹ (Human β-actin forward), and 5ʹ-AGCACTGTGTTGGCGTACAG-3ʹ (Human β-actin reverse).
Polyubiquitination assay
Polyubiquitination assays were performed as described previously26. Briefly, the 293 T cells were co-transfected with FLAG-DYRK1A-E396ter and HA-ubiquitin plasmids using the XtremeGene Transfection Reagent (Roche) for 24 h. Cells were then treated with 10 μM MG132 for 11 h, harvested, and lysed in an immunoprecipitation (IP) buffer (Tris/HCl, pH 7.5, 150 mM NaCl, 2 mM EDTA and 1% NP-40). The cell lysates were mixed with anti-FLAG M2 antibody-conjugated protein A/G-agarose beads (Santa Cruz Biotechnology) in 0.2% NP-40-containing IP buffer using a tubing-rotator at 4 °C for 2 h. The bound beads were washed three times with 0.2% NP-40-containing IP buffer and were solubilized with 2X sample buffer for western blot analysis.
Drosophilaculture
Drosophila melanogaster were cultured at 25 °C on standard cornmeal media. GMR-gal4 (#9146), MS1096-gal4 (#8860), Mhc-gal4 (#55133), UAS-Tau (human wild-type Tau, #51362)27, and all other stocks and balancers were obtained from the Bloomington Stock Centre (Bloomington, IN, USA). UAS-mnb transgenic flies were described in our previous report28. Based on amino acid sequence alignment, the human DYRK1A E396 residue corresponded to the Drosophila mnb D401 residue. Both wild-type full-length mnb and truncated mutant (mnb-D401ter) cDNAs were generated by PCR using Drosophila S2 cell cDNA as a template. Phusion Pfu PCR polymerase (Invitrogen) was used with the following primer pairs: 5ʹ-GAATTCATGTATAGATTAGAGGATACGA-3ʹ (forward, common for mnb-WT and mnb-D401ter), 5ʹ-TCTAGACTAATGTATAACTACAGGATTC-3ʹ (reverse for mnb-WT), and 5ʹ-TCTAGATCAGAAGAACTTGCGGGTCTTG-3ʹ (reverse for mnb-D401ter). The cDNAs of the full-length and truncated forms of mnb were ligated to the pUAST-FLAG vector digested with EcoRI/XbaI restriction enzymes to place the transgenes under the control of the UAS promoter. UAS-mnb-WT and UAS-mnb-D401ter transgenic flies were obtained by P-element-mediated germline transformation29. To analyse the eye phenotype, newly eclosed flies were collected and allowed to mate for 2–3 days. Wings from adult flies were dissected in 100% ethanol and mounted in Canada Balsam mounting medium (Gary’s magic mountant). The eye and wing phenotypes were photographed using a SZ60 binocular microscope equipped with an eXcope K5 CCD system (Olympus, Tokyo, Japan). Fly tissue sizes were measured on multiple samples (n > 10) from each genotype using NIH ImageJ software. The average eye size was presented as a normalized percentage of the control eye size. For adult lethality assessments, at least 100 flies were included in each individual experiment. For statistical analyses, two-tailed unpaired Student’s t-tests were used.
Immunohistochemistry
Immunostaining of larval samples was performed as previously described30. Briefly, fixed larval preparations were washed in phosphate buffered saline with Tween-20 (PBST) three times (10 min each), blocked in PBST with 5% normal goat serum for 30 min, and incubated with primary antibody at room temperature for 2 h or at 4 °C overnight. Fluorescein isothiocyanate-labelled goat anti-horseradish peroxidase (1:50 dilution, The Jackson Laboratory, Bar Harbour, ME, USA) was used to detect the synaptic bouton in larval tissue. All images were collected using a FV1000 confocal microscope (Olympus) and processed using NIH ImageJ software. Neuromuscular junction (NMJ) quantifications were conducted based on published procedures31. The filopodial NMJ structure was processed from complete z-stacks using the entire NMJ of the A4 abdominal segment. All statistical comparisons were performed using Prism software (GraphPad, San Diego, CA, USA). P-values were calculated using two-tailed Student’s t-tests.
Results
Case presentation
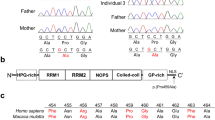
A 5-month-old female was referred to our hospital for evaluation of delayed development and facial dysmorphism. The patient was born after 39 weeks and 4 days of gestation by vaginal delivery after an uneventful pregnancy and weighed 2.89 kg. She was the third child of healthy Korean parents (Supplementary Fig. S1), and her siblings showed normal development. Her phenotypic features included an epicanthal fold, tented mouth, short and deep philtrum, deep-set eyes, bi-temporal narrowing, micrognathia, wide nasal bone, sparse scalp hair, and prominent ears with underdeveloped ear lobes (Supplementary Fig. S1). She also displayed rigidity in both her upper and lower extremities as well as thumb folding and deep tendon reflexes. An echocardiogram showed that she had a secondary atrial septal defect and peripheral pulmonary stenosis. Her chromosomal microarray examination revealed normal results. According to the Bayley Test (BSID-III), which measures early development in children, the patient had a language delay (composite score: 56; percentile score: 0.2), motor delay (composite score: 46; percentile score: <0.1), and poor adaptive behaviour skills (composite score: 66; percentile score: 1). At the time of her visit to the hospital, approximately 5 months after birth, her height was 63.6 cm (50th percentile), her weight was 5.8 kg (10th percentile), and her head circumference was 48.5 cm (25th percentile). Based on these clinical manifestations, the patient was initially suspected to have either Ohdo or Hallermann-Streiff syndrome. However, we discovered a novel heterozygous DYRK1A variant (c.1185dup, p.E396ter, [NM_001396.3]) on exon 8 by exome sequencing. The novel variant was only detected in the patient sample and was not present in the control population (variant databases from 1000 Genomes Project, ESP 6500, and ExAC). We confirmed the variant by Sanger sequencing (Fig. 1a). Parental segregation of the variant was negative, indicating that the mutation occurred in a sporadic form. Moreover, the patient’s brain MRI exhibited mild brain atrophy of both frontal lobes, thinning of the brainstem, hypoplasticity of the pituitary stalk and corpus callosum, and subcortical white matter hypomyelination, consistent with previously reported features of DYRK1A haploinsufficiency syndrome (Supplementary Fig. S1)21. After discovering that the patient’s disorder may be related to DYRK1A, rehabilitation therapy was initiated. At her 8-month follow-up (age: 13 months), the patient demonstrated delayed global developmental milestones and growth parameters. At 21 months of age, the patient developed seizures with generalized stiffness.

A novel de novo heterozygous mutation in the kinase domain of the DYRK1A gene. (a) Sanger sequencing confirmed a heterozygous novel DYRK1A variant, c.1185dup (p.E396ter) (NM_001396.3), in exon 8 that was identified by exome sequencing of the patient’s genome. (b) Schematic diagram of the DYRK1A catalytic domain. The location of the mutation and the translational termination codon (E396ter) is indicated as a red arrow.
Analysis of the structure and molecular dynamics of the DYRK1A-E396ter protein
The novel DYRK1A variant (c.1185dup) identified in this study was generated by the insertion of a single T at nucleotide position 1,185 of NM_001396.3. This insertion altered the translational frame, generated a premature translation termination codon (TGAG) at the E396 (GAG) position, and produced a truncated protein of 395 amino acids (DYRK1A-E396ter) (Fig. 1b). To gain a functional insight into the DYRK1A-E396ter protein, we primarily predicted its structure based on the crystal structure of the kinase domain in complex with its ATP-competitive inhibitor, DJM200532 (Protein Data Bank ID: 4MQ2) and substrate peptide (Fig. 2a). Structurally, the nonsense mutation occurred in the β-sheet of the CMGC insert, which only exists in CMGC kinases, thereby producing a protein lacking most of CMGC insert, α-helix H, and α-helix I at the C-terminal end of the catalytic domain. For the analysis of the molecular dynamics, we next performed energy minimization of the truncated structure by using the MODELLER software, and then the local molecular dynamics of the wild-type and E396ter DYRK1A proteins were assessed by using the Dynamut webserver with normal mode analysis function (Fig. 2b). As a result, we found that the DYRK1A-E396ter protein had elevated deformation energy throughout the protein compared to the wild-type one. Relatively high deformation energy was shown in a catalytic loop, an activation segment, and the loop between α8 and α9, suggesting a potential reduction of protein stability (Fig. 2c).

Analysis of the structure and molecular dynamics of the DYRK1A-E396ter protein. (a) Crystal structure of the human DYRK1A kinase domain in complex with its inhibitor, DJM2005 (PDB ID: 4MQ2) and substrate peptide is shown on the left. The C-helix, activation segment, catalytic loop, and CMGC insert are coloured in red, blue, orange, and hot pink, respectively. The N- and C-termini of the kinase domain of DYRK1A are represented by blue and red circles, respectively. The substrate peptide and inhibitor, DJM2005, are represented by a ball and stick model and coloured in dark and light grey, respectively. The predicted structure of DYRK1A-E396ter in complex with DJM2005 is shown on the right. The peptide sequences, which were not expressed in the mutant, are coloured dark grey and the location of the mutation (E396ter) is indicated by a red arrow. (b) Energy minimization of the wild-type and E396ter DYRK1A proteins was performed by using the MODELLER software, and the local molecular dynamics were assessed by using the Dynamut webserver with normal mode analysis function. Resulting molecular dynamics of the wild-type and E396ter DYRK1A proteins are presented in a tube style, which was generated by using PyMol software (version 1.3). The deformation energy is represented by thin to thick tubes coloured in blue (low), white (moderate), and red (high). (c) The deformation energy of the wild-type and E396ter DYRK1A proteins are coloured in blue and red, respectively.
DYRK1A-E396ter is efficiently degraded by the proteasome
To experimentally examine the protein stability of DYRK1A-E396ter in mammalian cells, we constructed a FLAG-tagged wild-type DYRK1A (FLAG-DYRK1A) expression clone and then introduced the mutation identified in the patient to produce a truncated protein of 395 amino acids (FLAG-DYRK1A-E396ter). In contrast to FLAG-DYRK1A-WT protein, FLAG-DYRK1A-E396ter protein was barely detectable by western blotting with an anti-FLAG antibody when overexpressed in 293 T cells (Fig. 3a). Since quantitative RT-PCR demonstrated that the transcripts were expressed at similar levels (Supplementary Fig. S2), we then examined the involvement of protein degradation. Protein degradation occurs in a specific and coordinated manner through major proteolytic systems, such as the proteasome, lysosome, calpains, and autophagy. To determine which systems contributed to the degradation of DYRK1A-E396ter, 293T cells expressing FLAG-DYRK1A-E396ter were treated separately with four selective inhibitors of each proteolytic degradation pathway. MG132, NH4Cl, calpeptin, and 3-methyladenine were used to inhibit proteasome-, lysosome-, calpain-, and autophagy-mediated protein degradation pathways, respectively. Western blotting with an anti-FLAG antibody revealed a prominent FLAG-DYRK1A-E396ter protein (~45 kDa) band only after treatment with MG132 (Fig. 3b) and not after treatment with any of the other inhibitors, suggesting that FLAG-DYRK1A-E396ter was degraded by the proteasome. Most proteins degraded by the proteasome are marked with (poly)ubiquitin chains. Thus, we analysed the ubiquitination of DYRK1A-E396ter by co-expressing FLAG-DYRK1A-E396ter and HA-ubiquitin in 293 T cells. Polyubiquitination of FLAG-DYRK1A-E396ter was observed at high levels when co-expressed with HA-ubiquitin and at even higher levels after additional treatment with MG132 (Fig. 3c). Together, these results indicate that DYRK1A-E396ter is degraded by the ubiquitin-dependent proteasomal degradation pathway.

DYRK1A-E396ter is degraded by the ubiquitin-mediated proteasomal pathway and is intrinsically inactive. (a) FLAG-tagged DYRK1A-WT, DYRK1A-E396ter, and DYRK1A-K188R plasmids were transiently co-expressed with Tau protein in 293 T cells for 24 h. Total cell extracts were harvested and subjected to western blotting with anti-FLAG, anti-phosphorylated-Tau (at T212, p-Tau), and anti-Tau antibodies. HnRNP A1 served as a loading control. Asterisks indicate nonspecific proteins. (b) FLAG-tagged DYRK1A-WT and DYRK1A-E396ter were co-expressed with Tau protein and then treated with selective proteolytic pathway inhibitors: MG132 for the proteasome, NH4Cl for lysosome, calpeptin for calpain, and 3-methyladenine for autophagy. (c) FLAG-DYRK1A-E396ter and HA-ubiquitin were co-expressed for 24 h prior to MG132 treatment (10 μM, 11 h). Total cell extracts were harvested, FLAG-DYRK1A-E396ter was immunoprecipitated with anti-FLAG antibody, and polyubiquitination was detected by western blotting with an anti-HA antibody. Uncropped full-sized blots are presented in Supplementary Fig. S4.
Loss-of-function of the DYRK1A patient mutant due to intrinsic inactivity
As the DYRK1A-E396ter protein was truncated at the catalytic domain, we sought to examine its kinase activity. We selected the Tau, a microtubule-associated protein, for the evaluation, because it is one of the most well-studied substrates of DYRK1A, and aberrant Tau phosphorylation is associated with the formation of neurofibrillary tangles in DS and Alzheimer’s disease10,33. Phosphorylation of Tau at the T212 residue is highly dependent on DYRK1A and was chosen as a marker of DYRK1A kinase activity34. Tau was transiently co-expressed with each FLAG-DYRK1A-WT, FLAG-DYRK1A-E396ter, and FLAG-DYRK1A-K188R1 (a catalytically inactive mutant of human DYRK1A) in 293 T cells. Phosphorylation of Tau at T212 was detected by western blotting with a phosphorylation-specific antibody. As previously reported, the co-expression of Tau and wild-type DYRK1A induced remarkable phosphorylation of Tau (Fig. 3a)27. In contrast, the DYRK1A-E396ter protein had little effect on Tau phosphorylation, which was similar to the DYRK1A-K188R protein.
We went on to examine whether FLAG-DYRK1A-E396ter possessed any intrinsic kinase activity when stabilized in the presence of MG132 (Fig. 3b). Despite a dramatic increase of FLAG-DYRK1A-E396ter protein level following the inhibition of proteasomal degradation, Tau phosphorylation was not rescued to an appreciable degree. These results indicate that the DYRK1A-E396ter mutant is catalytically inactive due to the truncation of the C-terminal region of the kinase domain. Additionally, we suggest that the intrinsic inactivity of DYRK1A-E396ter is the direct cause of its loss-of-function.
DYRK1A-E396ter does not have a dominant-negative effect on wild-type DYRK1A
Because both wild-type and mutant DYRK1A alleles are present in the patient’s cells, we investigated a potential dominant-negative effect of DYRK1A-E396ter on wild-type DYRK1A. The wild-type DYRK1A and DYRK1A-E396ter plasmids, alone or in a 1:1 combination, were co-transfected with the Tau plasmid, and the transfected cells were treated with MG132. As previously shown (Fig. 3a), Tau phosphorylation was strongly induced by the expression of wild-type DYRK1A. However, the additional co-expression of DYRK1A-E396ter did not impact Tau phosphorylation (Fig. 4). Similar results were observed even when the DYRK1A-E396ter protein was stabilized by MG132 treatment. Together, our findings indicate that DYRK1A-E396ter has little negative effect on wild-type DYRK1A with respect to Tau phosphorylation.

Effect of DYRK1A-E396ter on wild-type DYRK1A-induced Tau phosphorylation. DYRK1A-WT and DYRK1A-E396ter plasmids, alone or in a 1:1 combination, were co-transfected with the Tau plasmid for 24 h and then treated with MG132 (10 μM, 15 h). The subsequent procedure was identical to that described in the legend of Fig. 3a. Uncropped full-sized blots are presented in Supplementary Fig. S4.
Loss-of-function of the DYRK1A patient mutant in a Drosophila model
Because basic biological and neurological properties are highly conserved between human and Drosophila, Drosophila models are widely used to understand the molecular pathology of human diseases. Indeed, the Drosophila genome is nearly 75% homologous with human disease genes35, and the Drosophila minibrain (mnb) gene is 82% identical to its human homolog, DYRK1A36,37. Loss-of-function mutations in mnb result in reduced brain size with abnormal visual and olfactory behaviour due to defects in neurogenesis and brain development38, which recapitulates the microcephaly phenotype of DYRK1A haploinsufficiency in human patients. In addition, the tissue-specific overexpression of mnb also induces various phenotypic and neurological defects in central nervous system structure, consistent with various phenotypes of DS patients39.
To evaluate the functionality of the DYRK1A-E396ter protein in vivo, we generated transgenic Drosophila harbouring wild-type mnb (UAS-mnb-WT) or truncated mnb (UAS-mnb-D401ter). Based on sequence comparison, Drosophila D401 was equivalent to human E396 (Supplementary Fig. S3). For tissue-specific overexpression, transgenic mnb flies were crossed with flies expressing tissue-specific Gal4 drivers. The resulting expression of mnb-WT and mnb-D401ter proteins was analysed by western blotting of total protein extracts from transgenic flies (HS-Gal4 > UAS-mnb-WT or -D401ter) (Fig. 5a). Similar to our results in mammalian cells (Fig. 3a), the D401ter protein was expressed at much lower levels than the wild-type protein.

Loss-of-function of the mnb-D401ter mutant, the Drosophila equivalent of human DYRK1A-E396ter. (a) Transgenic flies ubiquitously overexpressing wild-type mnb or patient-derived truncated mnb (mnb-D401ter) were generated, and the expression of each mnb protein was analysed by western blotting with an anti-FLAG antibody. Asterisks indicate nonspecific proteins. Uncropped full-sized blots are presented in Supplementary Fig. S4. (b) Each mnb protein was specifically overexpressed in wing tissue using the MS1096-gal driver, and the resulting defects in L5 vein formation were analysed in adult flies. (c) Each mnb protein and/or human Tau was specifically overexpressed in the eye using the GMR-gal driver, and the resulting eye defects were analysed in adult flies. (d) Retinal surface areas were measured, and average eye sizes are presented as a percentage of the control. (e) Each mnb protein was overexpressed in muscle tissue using the Mhc-gal driver, and the resulting morphology of neuromuscular junctions in larval tissues were examined by visualization of the synaptic bouton. (f) The number of synaptic boutons was quantified and presented as a percentage of the control. (g) Each mnb protein was ubiquitously or neuro-specifically overexpressed using the Actin5- or elav-gal driver, respectively, and embryonic lethality was examined. Viability was presented as a percentage. Two-tailed Student’s t-tests were used to calculate P-values, which are depicted with an asterisk.
Next, the function of the D401ter protein was examined by comparing the phenotypes of mnb-D401ter flies with those of mnb-WT flies. Overexpression of mnb-WT in wing tissue using the MS1096-Gal4 driver showed shortening of the L5 vein in the adult wing, which we previously identified as one of the most prominent phenotypic defects27. In contrast, there was no recognizable phenotypic defect when the D401ter protein was overexpressed in the wing (Fig. 5b). Overexpression of human Tau in Drosophila is known to cause a severe eye degeneration phenotype40, which was worsened by co-expression with mnb-WT (Fig. 5c,d). Exacerbation of the eye degeneration phenotype is likely due to Tau hyperphosphorylation. In contrast, co-expression with mnb-D401ter had no additional effect.
As neuromuscular junction (NMJ) morphology of Drosophila larvae is used as a tool to assess neuronal synapse formation and integrity41, we analysed the effect of the D401ter mutation of mnb on NMJ morphology in transgenic flies. Overexpression of functional mnb-WT in the postsynaptic muscle tissue using mhc-Gal4 significantly reduced the number of NMJ synaptic boutons. A similar effect was observed by siRNA-mediated knockdown of endogenous mnb (Fig. 5e,f). Compared with these results, mnb-D401ter overexpression had minimal effect on the formation of the NMJ bouton. Moreover, ubiquitous (Actin5C-Gal4 > UAS-mnb-WT) or pan-neuronal (elav-Gal4 > UAS-mnb-WT) expression of mnb-WT caused severe embryonic lethality of 80–100%, whereas mnb-D401ter expression barely affected fly viability or any other phenotypes (Fig. 5g). Collectively, these results indicate that the mnb-D401ter mutation in Drosophila, which corresponds to the human DYRK1A-E396ter mutation, results in loss of kinase activity and function.
Discussion
In this study, we presented a patient with multiple congenital anomalies, including facial dysmorphism, developmental delay, and abnormalities in the cardiovascular system and brain structure (Supplementary Fig. S1). Exome sequencing and segregation analyses revealed that these phenotypic manifestations may be explained by a novel de novo heterozygous mutation of the DYRK1A gene. This novel mutation generated a translational termination codon and produced a C-terminally truncated protein (DYRK1A-E396ter) (Fig. 1a). Structurally, the DYRK1A-E396ter protein lacked the C-terminal end of the kinase domain, including the CMGC insert, α-helix H, and α-helix I. These regions, especially α-helices H and I, have been previously demonstrated to be required for the catalytic activity of DYRK1A. For instance, Arranz et al. showed that an R467ter DYRK1A mutant is completely inactive, and an F478 DYRK1A frame-shift mutant has less than 15% of the activity of wild-type DYRK1A. Their results indicate that even α-helix I, which is located at the C-terminal end of the kinase domain, is critically required for full kinase activity42. Consistent with this observation, we demonstrated that the DYRK1A-E396ter mutant was catalytically inactive. As this mutant protein was efficiently degraded by the proteasome and was barely detectable in mammalian cells, we evaluated its intrinsic kinase activity by inhibiting protein degradation with MG132. We found that even after protein stabilization, the DYRK1A-E396ter protein had little Tau-phosphorylating activity (Fig. 3b). According to an extensive analysis of DYRK1A missense mutants, lack of substrate phosphorylation activity is highly correlated with lack of tyrosine autophosphorylation42, which may be the case for the DYRK1A-E396ter protein.
More importantly, we further revealed that the mutant protein did not have a dominant-negative effect on the wild-type protein. Tau phosphorylation induced by the expression of the wild-type protein was barely affected by the additional expression of the mutant protein, even though the level of the mutant protein was recovered to the level of the wild-type protein by treatment with MG132 (Fig. 4). In the case of the two alleles are expressed in the patient’s heterozygous cells, this result indicates a lack of dominant-negative effect of the mutant protein, which would be the result of the combined contribution of two effects: reduced stability of the mutant protein and no interference of the mutant protein on the activities of the wild-type one. We further confirmed the lack of dominant-negative function of the mutant protein in Drosophila as an in vivo model. Transgenic mnb-D401ter flies, which have the Drosophila mutation equivalent to human DYRK1A-E396ter, had no recognizable phenotypic defects in the wings, eyes, and NMJs, and no severe embryonic lethality, whereas mnb-WT transgenic flies had severe phenotypic defects and lethality (Fig. 5). Collectively, these results clearly show that the DYRK1A-E396ter protein is not only catalytically inactive but is also completely non-functional in mammalian cells and fly models.
According to previous reports, the C-terminal end of the DYRK1A kinase domain is required for protein stability as well as kinase activity42,43,44. Thus, loss of this region in the DYRK1A-E396ter mutant may directly affect protein stability. Indeed, we observed that the mutant protein was efficiently degraded by the ubiquitin-mediated proteasomal pathway and was consequently undetected in mammalian cells (Fig. 3b,c). It has been previously reported that DYRK1A degradation is mediated by binding of the E3 ubiquitin ligase SCFβTrCP to the N-terminus of DYRK1A45. In addition, the CDC37/HSP90 chaperone has been suggested to regulate DYRK1A protein stability through interaction with its N-terminal lobe46. Whether these two mechanisms are associated with the degradation of the DYRK1A-E396ter protein needs further experimental validation.
In conclusion, we have identified a novel de novo DYRK1A nonsense mutation in a patient with DYRK1A haploinsufficiency syndrome. The mutation generates a C-terminally truncated protein at the β-sheet of the CMGC insert within the kinase domain that behaves as a loss-of-function mutant in both mammalian cell and Drosophila models.
References
Kentrup, H. et al. Dyrk, a dual specificity protein kinase with unique structural features whose activity is dependent on tyrosine residues between subdomains VII and VIII. J. Biol. Chem. 271, 3488–3495 (1996).
Himpel, S. et al. Identification of the autophosphorylation sites and characterization of their effects in the protein kinase DYRK1A. Biochem. J. 359, 497–505 (2001).
Yabut, O., Domogauer, J. & D’Arcangelo, G. Dyrk1A overexpression inhibits proliferation and induces premature neuronal differentiation of neural progenitor cells. J. Neurosci. 30, 4004–4014 (2010).
Chen, C. K. et al. Activity-dependent facilitation of Synaptojanin and synaptic vesicle recycling by the Minibrain kinase. Nat. Commun. 5, 4246 (2014).
Litovchick, L., Florens, L. A., Swanson, S. K., Washburn, M. P. & DeCaprio, J. A. DYRK1A protein kinase promotes quiescence and senescence through DREAM complex assembly. Genes Dev. 25, 801–813 (2011).
Laguna, A. et al. The protein kinase DYRK1A regulates caspase-9-mediated apoptosis during retina development. Dev. Cell 15, 841–853 (2008).
de Graaf, K. et al. The protein kinase DYRK1A phosphorylates the splicing factor SF3b1/SAP155 at Thr434, a novel in vivo phosphorylation site. BMC Biochem. 7, 7 (2006).
Di Vona, C. et al. Chromatin-wide profiling of DYRK1A reveals a role as a gene-specific RNA polymerase II CTD kinase. Mol. Cell 57, 506–520 (2015).
Wegiel, J., Gong, C. X. & Hwang, Y. W. The role of DYRK1A in neurodegenerative diseases. FEBS J. 278, 236–245 (2011).
Liu, F. et al. Overexpression of Dyrk1A contributes to neurofibrillary degeneration in Down syndrome. FASEB J. 22, 3224–3233 (2008).
Kim, E. J. et al. Dyrk1A phosphorylates alpha-synuclein and enhances intracellular inclusion formation. J. Biol. Chem. 281, 33250–33257 (2006).
Zhukareva, V. et al. Sporadic Pick’s disease: a tauopathy characterized by a spectrum of pathological tau isoforms in gray and white matter. Ann. Neurol. 51, 730–739 (2002).
Yin, X. et al. Dyrk1A overexpression leads to increase of 3R-tau expression and cognitive deficits in Ts65Dn Down syndrome mice. Sci. Rep. 7, 619 (2017).
Pozo, N. et al. Inhibition of DYRK1A destabilizes EGFR and reduces EGFR-dependent glioblastoma growth. J. Clin. Invest. 123, 2475–2487 (2013).
Altafaj, X. et al. Neurodevelopmental delay, motor abnormalities and cognitive deficits in transgenic mice overexpressing Dyrk1A (minibrain), a murine model of Down’s syndrome. Hum. Mol. Genet. 10, 1915–1923 (2001).
Ahn, K. J. et al. DYRK1A BAC transgenic mice show altered synaptic plasticity with learning and memory defects. Neurobiol. Dis. 22, 463–472 (2006).
Roubertoux, P. L. et al. Differential Brain, Cognitive and Motor Profiles Associated with Partial Trisomy. Modeling Down Syndrome in Mice. Behav. Genet. 47, 305–322 (2017).
Fotaki, V. et al. Dyrk1A haploinsufficiency affects viability and causes developmental delay and abnormal brain morphology in mice. Mol. Cell. Biol. 22, 6636–6647 (2002).
Raveau, M., Shimohata, A., Amano, K., Miyamoto, H. & Yamakawa, K. DYRK1A-haploinsufficiency in mice causes autistic-like features and febrile seizures. Neurobiol. Dis. 110, 180–191 (2018).
van Bon, B. W. et al. Disruptive de novo mutations of DYRK1A lead to a syndromic form of autism and ID. Mol. Psychiatry 21, 126–132 (2016).
Ji, J. et al. DYRK1A haploinsufficiency causes a new recognizable syndrome with microcephaly, intellectual disability, speech impairment, and distinct facies. Eur. J. Hum. Genet. 23, 1473–1481 (2015).
Matsumoto, N. et al. Possible narrowed assignment of the loci of monosomy 21-associated microcephaly and intrauterine growth retardation to a 1.2-Mb segment at 21q22.2. Am. J. Hum. Genet. 60, 997–999 (1997).
Oh, J. Y. et al. Identification of a Heterozygous SPG11 Mutation by Clinical Exome Sequencing in a Patient With Hereditary Spastic Paraplegia: A Case Report. Ann. Rehabil. Med. 40, 1129–1134 (2016).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424 (2015).
Choi, M., Baek, J., Han, S. B. & Cho, S. Facile analysis of protein-protein interactions in living cells by enriched visualization of the p-body. BMB Rep. 51, 526–531 (2018).
Choi, K. et al. Regulation of diacylglycerol acyltransferase 2 protein stability by gp78-associated endoplasmic-reticulum-associated degradation. FEBS J. 281, 3048–3060 (2014).
Kim, H. et al. A chemical with proven clinical safety rescues Down-syndrome-related phenotypes in through DYRK1A inhibition. Dis. Model. Mech. 9, 839–848 (2016).
Hong, S. H. et al. Minibrain/Dyrk1a regulates food intake through the Sir2-FOXO-sNPF/NPY pathway in Drosophila and mammals. PLoS Genet. 8, e1002857 (2012).
Rubin, G. M. & Spradling, A. C. Genetic transformation of Drosophila with transposable element vectors. Science 218, 348–353 (1982).
Lee, K. S. et al. Drosophila short neuropeptide F signalling regulates growth by ERK-mediated insulin signalling. Nat. Cell Biol. 10, 468–475 (2008).
Lee, S., Liu, H. P., Lin, W. Y., Guo, H. & Lu, B. LRRK2 kinase regulates synaptic morphology through distinct substrates at the presynaptic and postsynaptic compartments of the Drosophila neuromuscular junction. J. Neurosci. 30, 16959–16969 (2010).
Anderson, K. et al. Pyrido[2,3-d]pyrimidines: discovery and preliminary SAR of a novel series of DYRK1B and DYRK1A inhibitors. Bioorg. Med. Chem. Lett. 23, 6610–6615 (2013).
Johnson, G. V. & Hartigan, J. A. Tau protein in normal and Alzheimer’s disease brain: an update. J. Alzheimers Dis. 1, 329–351 (1999).
Woods, Y. L. et al. The kinase DYRK phosphorylates protein-synthesis initiation factor eIF2Bepsilon at Ser539 and the microtubule-associated protein tau at Thr212: potential role for DYRK as a glycogen synthase kinase 3-priming kinase. Biochem. J. 355, 609–615 (2001).
Reiter, L. T., Potocki, L., Chien, S., Gribskov, M. & Bier, E. A systematic analysis of human disease-associated gene sequences in Drosophila melanogaster. Genome Res. 11, 1114–1125 (2001).
Shindoh, N. et al. Cloning of a human homolog of the Drosophila minibrain/rat Dyrk gene from “the Down syndrome critical region” of chromosome 21. Biochem. Biophys. Res. Commun. 225, 92–99 (1996).
Aranda, S., Laguna, A. & de la Luna, S. DYRK family of protein kinases: evolutionary relationships, biochemical properties, and functional roles. FASEB J. 25, 449–462 (2011).
Tejedor, F. et al. minibrain: a new protein kinase family involved in postembryonic neurogenesis in Drosophila. Neuron 14, 287–301 (1995).
Degoutin, J. L. et al. Riquiqui and minibrain are regulators of the hippo pathway downstream of Dachsous. Nat. Cell. Biol. 15, 1176–1185 (2013).
Jackson, G. R. et al. Human wild-type tau interacts with wingless pathway components and produces neurofibrillary pathology in Drosophila. Neuron 34, 509–519 (2002).
Frank, C. A. Homeostatic plasticity at the Drosophila neuromuscular junction. Neuropharmacology 78, 63–74 (2014).
Arranz, J. et al. Impaired development of neocortical circuits contributes to the neurological alterations in DYRK1A haploinsufficiency syndrome. Neurobiol. Dis. 127, 210–222 (2019).
Kornev, A. P., Taylor, S. S. & Ten Eyck, L. F. A helix scaffold for the assembly of active protein kinases. Proc. Natl. Acad. Sci. USA 105, 14377–14382 (2008).
Widowati, E. W., Ernst, S., Hausmann, R., Muller-Newen, G. & Becker, W. Functional characterization of DYRK1A missense variants associated with a syndromic form of intellectual deficiency and autism. Biol. Open 7, bio032862 (2018).
Liu, Q. et al. E3 Ligase SCFbetaTrCP-induced DYRK1A Protein Degradation Is Essential for Cell Cycle Progression in HEK293 Cells. J. Biol. Chem. 291, 26399–26409 (2016).
Sonamoto, R. et al. Identification of a DYRK1A Inhibitor that Induces Degradation of the Target Kinase using Co-chaperone CDC37 fused with Luciferase nanoKAZ. Sci. Rep. 5, 12728 (2015).
Acknowledgements
We thank the patient and her family for taking part in the study. This research was supported by grants funded by the National Research Foundation of Korea (NRF-2015M3A9C7030128, NRF-2019R1F1A1058972, and NRF-2020R1A2C2012347), UST Young Scientist Research Program through the University of Science and Technology (2018YS06R), and grants from the KRIBB Research Initiative Programs.
Author information
Authors and Affiliations
Contributions
C.K.C. and S.C. conceived project and supervised all procedure of this study. C.K.C., S.C., and K.S.L. designed experiments and analyzed data. M.C., D.W.K., D.K., J.M.C., A.K.K. and Y.H. performed the experiments and preparing the figures. D.K., J.M.C., and S.B.H. provided various intellectual and material support. C.K.C., S.C., K.S.L., M.C., D.K., and J.M.C. prepared figures and wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
Written and informed consent was obtained from the parents of the patient for the publication of this manuscript and the accompanying images of the patient. A copy of the patient’s consent is accessible to the Editor of this journal. The authors have no conflicting interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lee, KS., Choi, M., Kwon, DW. et al. A novel de novo heterozygous DYRK1A mutation causes complete loss of DYRK1A function and developmental delay. Sci Rep 10, 9849 (2020). https://doi.org/10.1038/s41598-020-66750-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-66750-y
This article is cited by
-
Epigallocatechin gallate attenuates neurocognitive impairment in CRISPR-CAS9-induced knockout of DYRK1A gene in zebrafish model
Advances in Traditional Medicine (2023)
-
Characterizing Sensory Phenotypes of Subgroups with a Known Genetic Etiology Pertaining to Diagnoses of Autism Spectrum Disorder and Intellectual Disability
Journal of Autism and Developmental Disorders (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
