
Abstract
Modulation of tumor microenvironment is an emerging frontier for new therapeutics. However in meningiomas, the most frequent adult brain tumor, the correlation of microenvironment with tumor phenotype is scarcely studied. We applied a variety of systems biology approaches to bulk tumor transcriptomics to explore the immune environments of both skull base and convexity (hemispheric) meningiomas. We hypothesized that the more benign biology of skull base meningiomas parallels the relative composition and activity of immune cells that oppose tumor growth and/or survival. We firstly applied gene co-expression networks to tumor bulk transcriptomics from 107 meningiomas (derived from 3 independent studies) and found immune processes to be the sole biological mechanism correlated with anatomical location while correcting for tumour grade. We then derived tumor immune cell fractions from bulk transcriptomics data and examined the immune cell-cytokine interactions using a network-based approach. We demonstrate that oncolytic Gamma-Delta T cells dominate skull base meningiomas while mast cells and neutrophils, known to play a role in oncogenesis, show greater activity in convexity tumors. Our results are the first to suggest the importance of tumor microenvironment in meningioma biology in the context of anatomic location and immune landscape. These findings may help better inform surgical decision making and yield location-specific therapies through modulation of immune microenvironment.
Similar content being viewed by others


Introduction
Meningiomas are amongst the most common adult brain tumors and constitute approximately 30% of all intracranial neoplasms1. Surgery remains a key part of treatment for symptomatic or growing meningiomas and outcomes are largely determined by tumor biology2,3 and extent of resection4. Approximately 70% of meningiomas have benign characteristics and the average disease-free survival is 90% over 10 years2, but lifetime recurrence can be as high as 50%5. The extent of surgical excision is largely determined by technical feasibility which is a function of anatomical location of the tumor, adherence to adjacent tissue and the eloquent structures limiting the extent of removal6. Achieving complete excision of meningiomas that grow around structures at the base of the skull (so-called “skull base meningiomas”) poses a particular challenge due to the proximity of neurovascular structures as well as the often narrow surgical corridors (these are contrasted from “convexity meningiomas” which grow elsewhere in the supratentorial space). Consequently, they may require longer operative times and can have lower rates of complete excision. Given the increased likelihood of being left with post-operative residual tumour, understanding the biology skull base meningiomas is of particular interest when it comes to developing new medical treatments to prevent further growth or recurrence.
Multiple studies show that skull base meningiomas are more likely to have benign biology whereas tumors with more aggressive behavior (atypical or malignant meningiomas) can constitute close to 30% of convexity/parafalcine tumors7,8,9. Recent studies have analyzed the genetic makeup of meningiomas and found recurrent mutations in the neurofibromatosis type 2 (NF2) gene and/or loss of chromosome 22 (NF2/chr22loss) to be more prevalent in the cerebral and cerebellar hemisphere10. The vast majority of non-NF2/chr22loss meningiomas are typically benign and tend to be located medially on the skull base. Smoothened, frizzled family receptor (SMO) mutations are also implicated in non-NF2/chr22loss and medial meningiomas through increased activation of the Hedgehog pathway10. Recurrent polymerase (RNA) II (DNA directed) polypeptide A (POLR2A) mutations have recently been found to classify a distinct subset of benign meningiomas with meningothelial histology and a preponderance to localize in the tuberculum sellae11. Analysis of methylation subtypes of meningiomas showed a relatively greater proportion of skull base lesions among the more benign subgroups with longer disease-free survival, when compared to the groups with less favourable outcomes. On the other hand, all tumors in the malignant methylation subtype were located exclusively on the convexity8.
Systems level analysis of gene expression data, which considers genes as units of a system rather than isolated entities, can identify key molecular processes which map to biological/clinical phenotypes12,13. This approach carries the distinct advantage of being unbiased in deriving meaning from high dimensional, biologically-modelled data. Gene co-expression networks are one example of such system level analysis, wherein similar genes are grouped into “modules” of similar function14. This technique has been successfully applied to explore complex phenotypes in Huntington’s disease15, peripheral nerve regeneration16 and weight gain17. It has also been used to identify relevant subgroups of meningioma18. In the current study we apply this technique along with another network-based approach to model the immune landscape of skull base and convexity meningiomas.
We hypothesize that different meningioma locations are associated with distinct biological mechanisms, likely related to immune cell composition and/or activity. To capture these geographical patterns in meningioma biology we analyzed the transcriptomics profiles of 107 meningiomas originating from either the skull base or convexity.
Results
Co-expression network and module-phenotype correlation
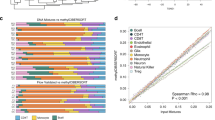
A gene co-expression network was constructed from 19,011 gene transcripts. Using an adaptive hierarchical clustering model14, we discovered fourteen gene modules (Fig. 1A). We next compared the biology of both tumor locations by correlating phenotype with meta-gene expression levels. Three modules were found to be significantly different between hemispheric and skull base meningiomas (Mann Whitney p < 0.05), only one of which annotated significantly (Bonferroni p < 0.05) to DAVID gene ontology/pathways (Fig. 1B,C). We do note that one of these modules mapped significantly to the “extracellular exosome” cellular compartment, but did not annotate to anything else in DAVID and therefore was not considered in further analysis. We also confirmed module significance by correcting the model for WHO grade, age, and sex and note that significance is preserved for all 3 modules. Finally, we selected the 10 most correlated genes to the module eigengene from the 3 modules which are significantly associated with tumour location and used these 30 genes in a logistic regression model to predict tumour location. The area under the resultant receiver-operating characteristic (ROC) curve was 0.849 (Supplemental Fig. 2).

Gene co-expression network reveals immune function to correlate strongly with meningioma location. (A) Gene dendrogram illustrating modules. The grey denotes genes which are not implicated with any modules. Labels indicate cytokines which are positively correlated with the module meta-gene expression (Pearson correlation>0.6, p < 0.05). (B) Boxplot depicting the correlation between location and meta-gene expression level of the only significant module with DAVID annotations (Mann Whitney p = 0.005). As indicated in (C), this module maps to diverse immune processes, and is therefore labeled as “immune response (universal)”. C: Gene ontology terms ranked by Bonferroni p-value.
Immune composition of meningiomas by location
Using network analysis, we examined the biological activity of each immune cell type by correlating them with cytokine expression. On inspection of cytokine-cell networks there was a clear difference in the network configurations of convexity and skull base meningiomas (Fig. 2). Notably, activated mast cells and neutrophils are present in the convexity network with our cutoff of Pearson ρ>0.6, p < 0.05. In this network, mast cells are most correlated with IL-6 (ρ = 0.69, p = 3.0 ×10−6) and neutrophils with IL1R2 (ρ = 0.74, p = 2.4 ×10−7). In the skull base network, there are no cell-ligand interactions that meet our threshold, though we note that the strongest associations are between BTK and both plasma cells (ρ = 0.57, p = 2.4 ×10−7) and monocytes (ρ = 0.54, p = 1.3 ×10−6).

Network demonstrating the connectivity of immune cell fractions and cytokine transcriptomics in convexity (A) and skull base (B) meningiomas. Cytokines are represented in blue whereas immune cells are represented in red. Edge thickness is proportional to Pearson correlation and node size is proportional to eigenvector centrality, a measure of the influence of a particular node in the network. Notably, the eigenvector centrality ranges from 0 to 0.39 (IL-6) in the convexity model and from 0 to 0.50 (CCL3) in the skull base model. Neutrophils and activated mast cells are significantly correlated with cytokines in the convexity model using our thresholding criteria, while the skull base model contains only cytokines.
Given the relative complexity of these networks, we sought to analyze the relative “activity” of each immune cell by probing the distribution of their correlations with cytokine expression levels19. Analysis of cell “connectivity” (the area under this distribution) as well as eigenvector centrality (an established measure of node influence within a network) demonstrates the importance of mast cells and neutrophils in convexity meningiomas and of gamma-delta T cells, monocytes, and plasma cells in skull base meningiomas (Fig. 3).

Immune cell connectivity of meningiomas by location. A-B: Histogram of Person’s correlations for the top 3 cell fractions, ranked by connectivity, of convexity (A) and skull base (B) meningiomas. C: Difference between cell connectivity values, comparing convexity to skull base (“SB”), where positive values (blue) indicate greater connectivity in convexity while negative values (red) indicate greater connectivity in skull base meningiomas. D and E: Ranking of eigenvector centrality of each cell type for convexity (D) and skull base (E) tumours. F: Difference between eigenvector centrality, with the same conventions as (C). Note the highly connected mast cells and neutrophils in convexity meningiomas and T gamma-delta cells, monocytes, and plasma cells in skull base (“SB”) meningiomas. A = activated, M = mature, N = naïve, fh = follicular helper, gd = gamma-delta, R = resting.
Discussion
We present a genetic meta-analysis correlating transcriptomics profile with meningioma location. Using a systems biology approach, we demonstrate an upregulation of various immune processes in skull base meningiomas compared to convexity (hemispheric) meningiomas. We further investigated the immune landscape of meningiomas by combining leukocyte cell fraction with cytokine expression profile in a network-based analysis. Gamma delta T cells were associated with the greatest cytokine connectivity and eigenvector centrality in our network of skull base meningiomas, while mast cells and neutrophils played the most central roles in the convexity network. This finding may explain the more benign biology of skull base meningiomas when compared to convexity meningiomas.
It is known that the more aggressive grade II and grade III meningiomas are less prevalent in the skull base compared to convexity meningiomas7,20. When comparing to meningiomas of the convexity using gene co-expression analysis, a key immune module showed significant upregulation in meningiomas of the skull base which was maintained after correcting for WHO grade. This suggests that cellular microenvironment, and in particular the immune microenvironment, may play an important role in modulating tumor behaviour and in explaining differences observed between the two subsets of meningioma being studied. The important role of tumour microenvironment has been well demonstrated in a variety of pan-cancer12,21 and central nervous system tumour studies22, and has been suggested in meningiomas23,24,25. Additionally, multiparameter flow cytometry studies have demonstrated a heterogenous composition of immune cells in meningiomas23,25. In our study, Gamma delta T cells were amongst the most active cell fraction in the skull base cohort based on both overall cytokine correlation and eigenvector centrality. Notably, this cell fraction is regarded as an inhibitor of tumor growth26. We also note importance of plasma cells and monocytes in this cohort, whose roles are less clear. Interestingly, tumour-infiltrating plasma cells have been associated with anti-tumoral activity in ovarian cancer27, and monocytes are noted to have both pro and anti-tumoral effects in cancer28. In convexity meningiomas, by contrast, we identified activated mast cells and neutrophils as the most connected fraction. Tumor-associated mast cells may support the oncogenic environment by releasing pro-tumorigenic stimulants29, thereby triggering angiogenesis, tumor cell proliferation/invasion, formation of lymphatic/blood vessel and facilitate the process of extravasation of cytokine-producing cells. Mast cells have been detected in upto 90% of high-grade meningioma tissue and has been also found to correlate with peritumoral edema30, a feature of aggressive biology in these tumours. Additionally, tumour-associated neutrophils have been implicated in cancer progression and poor clinical outcomes31.
Conventional analysis of transcriptomics data often relies on differential single-gene expression levels to filter out relevant patterns by comparing the “diseased” and “normal” tissue. These techniques may overlook relatively subtle effects across several highly-connected genes, which may nevertheless trigger a robust cellular mechanism. Gene expression networks are well suited for detecting small, additive biological signals that underlie relevant clinical phenotypes. In the first part of our analysis we used this well-established technique14 to show that immune processes correlate with location of meningiomas, and investigated this finding further using a network-based representation of the interactions between immune cell fractions and cytokine expression patterns. In this second technique, we used the sum of cell-cytokine correlations as a measure of immune cell “activity”, which is inspired by existing network based approaches19. Importantly, this output corresponds well to eigenvector centrality. Although further verification of this approach is needed, we highlight distinct cell fractions and interactions between skull base and convexity meningiomas, which align with known differences in clinical behaviour.
There are a number of limitations to this research which must be considered. Firstly, follow up data is lacking from all series we have includes which limits our ability to comment on tumour aggressiveness or recurrence rates, though our conclusions are nevertheless in alignment with prior knowledge on tumor immunology and the biology of convexity vs skull base meningiomas. We also do not have data on treatment regimens at the time of tissue collection, though as all patients undergo surgery for primary (non-recurrent) meningiomas and the vast majority of tumours our cohort are WHO grade 1, it’s very unlikely that any patients received neo-adjuvant treatment. Additionally, the proportion of grade II meningiomas is relatively low in our study (5.3%) compared to the prevalence in the population (20–30%). To address this, we have corrected for grade when correlating module gene expression with location. We also acknowledge that since skull base tumours are likely to produce symptoms and therefore be treated earlier, this may have an effect on the differences in observed biology. Notably, the case series of Magill et al.7 shows mean diameters of 2.6–3.6 cm in the skull base compared to 3.6–4.5 cm in convexity/falcine/parasaggital regions at the time of surgical excision. However, these findings are likely due to the well documented differences in genetic makeup8,10, biology, and growth rate of meningiomas. In non-skull base meningiomas, growth rate is higher and time to doubling is lower compared to skull base meningiomas32. Furthermore, the transformation of meningioma biology over time is quoted at a rate of 1–2%33 suggesting that the distinct biology is likely present from the initial occurrence of the meningioma rather than acquired over time. Consequently, the time of discovery/resection is unlikely to play a significant role. Finally, our assessment of cytokine activity is on the transcriptomics level as no secretory data was available, yet this approach still provides an indirect assessment of how “active” an immune cell type is in terms of cytokine synthesis. Additionally, we note that transcriptomics have been successfully used to infer autocrine and heterocrine cellular functions with good correlation between mRNA abundance and downstream protein synthesis. Such approaches have been successfully implemented to infer cell-cell interactions in lung development34 and aging of the mouse brain35. Importantly, we have been able to identify a robust, recurrent signal despite considerable data heterogeneity which lends itself to further confirmatory testing with biological assays.
Conclusions
Our study is the first to computationally estimate immune cell fractions in location-specific meningioma tissue from bulk transcriptomics. We demonstrate distinct immune compositions between hemispheric and skull base meningiomas using a network-based approach that considers cell connectivity with cytokine transcriptomics. Gamma-delta T cells, with potentially tumour-suppressant activity, are more dominant in skull base meningiomas which is in keeping with a more benign biology. This is in contrast to the more central role of potentially oncogenic mast cells and neutrophils in convexity meningiomas. These findings give further insight into the immune microenvironment of meningiomas and may have implications on future strategies of immune modulation for this challenging disease.
Methods
Data preparation
All data was collected from the Gene Expression Omnibus (GEO), a public repository of high-throughput functional genomic data sets36. We used studies containing details on meningioma location (skull base and convexity) and WHO grade with corresponding gene expression data11,37, providing us with a cumulative sample size of 107 meningiomas (Table 1). All studies obtained microarray data from surgically excised primary meningioma tissue. Summary statistics for each study are presented in Table 1. We note that a diagram outlining the workflow can be found in Supplemental Fig. 1.
Pre-processing of transcriptomics data
For each study, the microarray data was backgrounded corrected, quantile normalized, and log-2 transformed using the Affy39 and Limma40 R packages for Affymetrix and Illumina platforms, respectively. After removing genes that were not common across these studies (such that the cohorts could be merged into a single matrix) we were left with 19,011 genes. The 3 studies were then merged, scaled to a global mean and standard deviation of 0 and 1, respectively41, and batch-corrected using ComBat, a well-established empirical Bayes approach42. The resultant data matrix was used during all subsequent analysis.
Co-expression network analysis
We performed WGCNA of gene expression data using R (version 3.5.1) to construct a co-expression network and identify biological modules which map to meningioma location. Pairwise gene correlations were soft-thresholded with an exponent of 20 to approximate scale-free topology, which was ultimately transformed into a biologically-inspired “Topological Overlap Matrix” (TOM), which measures pairwise gene similarity in terms of shared topology within the full network14. Highly similar genes are then grouped into an adaptive hierarchical clustering tree (dendrogram), yielding “modules” with a minimum size of 30 genes. The gene expression profile of each module is represented by a meta-gene computed as its first principal component, an established method. The meta-gene score for each module is then compared between skull base and convexity meningiomas, with significance considered at Mann Whitney p < 0.05. As an exploration of the relationship between cytokine activity and module expression, we label cytokines whose expression levels are significantly positively associated with module meta-gene expression (Pearson correlation>0.6, p < 0.05). Finally, to assess for network robustness, we select the 10 most correlated genes to the module eigengene from all modules which are significantly associated with tumour location and use their expression as inputs into a logistic regression model to predict location.
Module-based qualitative analysis
Genes in each of the identified modules were annotated by the Database for Annotation, Visualization and Integrated Discovery (DAVID version 6.8)43.
Deconvolution of tumor bulk expression signal
We used CIBERSORT44 (Package EpiDISH version 1.4.1), a well-established technique to estimate immune cell fractions from our gene expression data. Briefly, the method uses support vector regression to compare bulk transcriptomic data from previously derived signature matrices from 22 different immune cells in order to estimate the relative prevalence of each of these 22 immune cells in the bulk data. The specifics of this technique are beyond the scope of this paper and are described in detail in the attached reference. Notably, the input gene expression data is not log transformed and is scaled to a global mean and standard deviation of 0 and 1, respectively.
Network analysis of immune cell fractions and expression of cytokines
The complex relationships between immune cells and cytokine expression levels were visualized using network analysis, which demonstrates associations that are otherwise difficult to appreciate. Such network-based approaches have revolutionized research into phenom-genotype similarities and elucidated the genetic basis of diseases45,46, predicted pathways of disease progression19, and identified novel drug targets47. Nodes represent immune cells (CIBERSORT output) and list of 35 known cytokines derived from the literature21,48 (Supplemental data). Edges represent Pearson correlations, wherein a significance cut-off of <0.05 and effect size cutoff of>0.6 are used. Node size is proportional to the eigenvector centrality as computed by the CYTOSCAPE (version 3.6.1) interface. We also compute an unweighted eigenvector centrality score (a measure of node influence within a network which is independent of Pearson correlations) for each cell types using a network wherein adjacency is defined by significant correlations (p < 0.05) in order to assess the degree of influence of each cell within each network. We also define “connectivity” as the biological activity of each immune cell (i.e. its overall expression of cytokines), computed as the sum of all significant (p < 0.05) Pearson correlations between a cell type and the expression levels of 35 pre-determined cytokines (Supplementary data). Cell types were ranked based on both of these metrics.
Data availability
All data referenced in this study is publicly available through the open repository Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/).
References
Wiemels, J., Wrensch, M. & Claus, E. B. Epidemiology and etiology of meningioma. J. Neurooncol. 307–314 (2010). https://doi.org/10.1007/s11060-010-0386-3.
Rogers, L. et al. Meningiomas: knowledge base, treatment outcomes, and uncertainties. A RANO review. J. Neurosurg. 122, 4–23 (2015).
Goldbrunner, R. et al. EANO guidelines for the diagnosis and treatment of meningiomas. Lancet Oncol. 17, e383–e391 (2016).
Sughrue, M. E. et al. The relevance of Simpson Grade I and II resection in modern neurosurgical treatment of World Health Organization Grade I meningiomas. J. Neurosurg. 113, 1029–1035 (2010).
Jääskeläinen, J. Seemingly complete removal of histologically benign intracranial meningioma: Late recurrence rate and factors predicting recurrence in 657 patients. A multivariate analysis. Surg. Neurol. 26, 461–469 (1986).
Demonte, F., McDermott, M. W. & Al-mefty, O. Al-Mefty’s Meningiomas. (2011).
Magill, S. T. et al. Relationship between tumor location, size, and WHO grade in meningioma. Neurosurg. Focus https://doi.org/10.3171/2018.1.FOCUS17752 (2018).
Sahm, F. et al. DNA methylation-based classification and grading system for meningioma: a multicentre, retrospective analysis. Lancet Oncol. 18, 682–694 (2017).
Liang, R. F. et al. The potential risk factors for atypical and anaplastic meningiomas clinical series of 1,239 cases. Int. J. Clin. Exp. Med. (2014).
Clark, V. E. et al. Genomic analysis of non-NF2 meningiomas reveals mutations in TRAF7, KLF4, AKT1, and SMO. Science (80-.). 339, 1077–1080 (2013).
Clark, V. E. et al. Recurrent somatic mutations in POLR2A define a distinct subset of meningiomas. Nat. Genet. 48, 1253–1259 (2016).
Thorsson, V. et al. The Immune Landscape of Cancer. Immunity 48, 812–830.e14 (2018).
Hawrylycz, M. et al. Canonical genetic signatures of the adult human brain. Nat. Neurosci. 18, 1832–1844 (2015).
Langfelder, P. & Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinformatics 9, (2008).
Langfelder, P. et al. Integrated genomics and proteomics define huntingtin CAG length-dependent networks in mice. Nat. Neurosci. 19, 623–633 (2016).
Chandran, V. et al. A Systems-Level Analysis of the Peripheral Nerve Intrinsic Axonal Growth Program. Neuron 89, 956–970 (2016).
Ghazalpour, A. et al. Integrating genetic and network analysis to characterize genes related to mouse weight. PLoS Genet. 2, 1182–1192 (2006).
Zador, Z. et al. Authors: BioRxiv Homogenous subgroups of atypical meningiomas defined using oncogenic signatures: (2018).
Hidalgo, A. & Blumm, N. A Dynamic Network Approach for the Study of Human Phenotypes. PLoS Comput. Biol. 5, (2009).
Mansouri, A. et al. Surgically resected skull base meningiomas demonstrate a divergent postoperative recurrence pattern compared with non–skull base meningiomas. J. Neurosurg. https://doi.org/10.3171/2015.7.JNS15546 (2016).
Binnewies, M. et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 24, 541–550 (2018).
Lim, M., Xia, Y., Bettegowda, C. & Weller, M. Current state of immunotherapy for glioblastoma. Nat. Rev. Clin. Oncol. 1–21 https://doi.org/10.1038/s41571-018-0003-5 (2018).
Fang, L. et al. The immune cell infiltrate populating meningiomas is composed of mature, antigen-experienced T and B cells. Neuro. Oncol. 15, 1479–1490 (2013).
Pinton, L. et al. Immunosuppressive activity of tumor-infiltrating myeloid cells in patients with meningioma. Oncoimmunology 7, (2018).
Domingues, P. H. et al. Immunophenotypic identification and characterization of tumor cells and infiltrating cell populations in meningiomas. Am. J. Pathol. 181, 1749–1761 (2012).
Pauza, C. D., Liou, M., Lahusen, T., Xiao, L. & Lapidus, R. G. Gamma Delta T Cell Therapy for Cancer: it is Good to be Local. Front. Immunol. 9, (2018).
Kroeger, D. R., Milne, K. & Nelson, B. H. Tumor-Infiltrating Plasma Cells Are Associated with Tertiary Lymphoid Structures, Cytolytic T-Cell Responses, and Superior Prognosis in Ovarian Cancer. Clin. Cancer Res. 1–12 https://doi.org/10.1158/1078-0432.CCR-15-2762 (2016).
Olingy, C. E., Dinh, H. Q. & Hedrick, C. C. Monocyte heterogeneity and functions in cancer. J. Leukoc. Biol. 309–322 https://doi.org/10.1002/JLB.4RI0818-311R (2019).
Aponte-López, A., Fuentes-Pananá, E. M., Cortes-Muñoz, D. & Muñoz-Cruz, S. Mast Cell, the Neglected Member of the Tumor Microenvironment: Role in Breast Cancer. J. Immunol. Res. 2018, (2018).
Polyzoidis, S., Koletsa, T., Panagiotidou, S., Ashkan, K. & Theoharides, T. C. Mast cells in meningiomas and brain inflammation. J. Neuroinflammation 12, 1–8 (2015).
Shaul, M. E. & Fridlender, Z. G. Tumour-associated neutrophils in patients with cancer. Nat. Rev. Clin. Oncol. https://doi.org/10.1038/s41571-019-0222-4 (2019).
Hashimoto, N. et al. Slower growth of skull base meningiomas compared with non–skull base meningiomas based on volumetric and biological studies. J. Neurosurg. 116, 574–580 (2012).
Lamszus, K. et al. Allelic Losses at 1p, 9q, 10q, 14q, and 22q in the Progression of Aggressive Meningiomas and Undifferentiated Meningeal Sarcomas. Cancer Genet. Cytogenet. 4608, (1999).
Raredon, M. S. B. et al. Single-cell connectomic analysis of adult mammalian lungs. Sci. Adv. 2, 1–15 (2019).
Ximerakis, M. et al. Single-cell transcriptomic profiling of the aging mouse brain. Nat. Neurosci. 22, (2019).
Barrett, T. et al. NCBI GEO: archive for functional genomics data sets — update. Nucleic Acids Res. 41, 991–995 (2013).
Dalan, A. B. et al. Simultaneous analysis of miRNA-mRNA in human meningiomas by integrating transcriptome: A relationship between PTX3 and miR-29c. BMC Cancer 1–9 https://doi.org/10.1186/s12885-017-3198-4 (2017).
Schulten, H., Hussein, D., Al-adwani, F. & Karim, S. Microarray Expression Data Identify DCC as a Candidate Gene for Early Meningioma Progression. PLoS One 1–17 https://doi.org/10.1371/journal.pone.0153681 (2016).
Gautier, L., Cope, L., Bolstad, B. M. & Irizarry, R. A. Affy - Analysis of Affymetrix GeneChip data at the probe level. Bioinformatics 20, 307–315 (2018).
Ritchie, M. E. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, (2015).
Hughey, J. J. & Butte, A. J. Robust meta-analysis of gene expression using the elastic net. Nucleic Acids Res. 43, 1–11 (2015).
Chen, C. et al. Removing Batch Effects in Analysis of Expression Microarray Data: An Evaluation of Six Batch Adjustment Methods. PLoS One 6, (2011).
Huang, D. W., Sherman, B. T. & Lempicki, R. A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57 (2009).
Newman, A. M. et al. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 12, 453–457 (2015).
Califano, A., Butte, A. J., Friend, S., Ideker, T. & Schadt, E. Leveraging models of cell regulation and GWAS data in integrative network-based association studies. Nat. Publ. Gr. 44, 841–847 (2012).
Scicluna, B. P. et al. Van Der. Modular Transcriptional Networks of the Host Pulmonary Response during Early and Late Pneumococcal Pneumonia. Mol. Med. 3, 430–441 (2015).
Barabási, A. L., Gulbahce, N. & Loscalzo, J. Network medicine: A network-based approach to human disease. Nat. Rev. Genet. 12, 56–68 (2011).
Xiao, W. et al. A genomic storm in critically injured humans. J Exp Med 208, 2581–2590 (2011).
Acknowledgements
The authors would like to thank Dr. Claudia Dos Santos and Dr. Christopher Walsh for their assistance with analytic techniques.
Author information
Authors and Affiliations
Contributions
Z.Z.: study design, manuscript preparation. A.P.L.: data analysis, manuscript revisions. M.B.: data analysis, manuscript revisions. M.D.C.: manuscript revisions
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zador, Z., Landry, A.P., Balas, M. et al. Landscape of immune cell gene expression is unique in predominantly WHO grade 1 skull base meningiomas when compared to convexity. Sci Rep 10, 9065 (2020). https://doi.org/10.1038/s41598-020-65365-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-65365-7
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
