
Article
|
Open Access
Featured
-
-
Article
| Open Access Extreme purifying selection against point mutations in the human genome
Extreme purifying selection against point mutations in the human genome
Previous work has investigated selection in the coding genome, but it is not as well characterized in the non-coding genome. By analyzing rare variants in 70k genome sequences from gnomAD, the authors detect very strong purifying selection ("ultraselection”) across the human genome, finding it in some microRNAs and coding sequences but generally rare in regulatory sequences.
- Noah Dukler
- , Mehreen R. Mughal
- & Adam Siepel
-
Article
| Open Access KSTAR: An algorithm to predict patient-specific kinase activities from phosphoproteomic data
KSTAR: An algorithm to predict patient-specific kinase activities from phosphoproteomic data
Kinases are important drug targets, but predicting their activities from phosphoproteomics data remains challenging. While many existing prediction tools rely on phosphosite-specific quantitative data, Crowl et al. develop a kinase activity prediction algorithm that requires no phosphosite quantification.
- Sam Crowl
- , Ben T. Jordan
- & Kristen M. Naegle
-
Article
| Open Access An analysis of 45 large-scale wastewater sites in England to estimate SARS-CoV-2 community prevalence
An analysis of 45 large-scale wastewater sites in England to estimate SARS-CoV-2 community prevalence
Wastewater surveillance could provide a means of monitoring SARS-CoV-2 prevalence that does not rely on testing individuals. Here, the authors report results from England’s national wastewater surveillance program, use it to estimate prevalence, and compare estimates with those from population-based prevalence surveys.
- Mario Morvan
- , Anna Lo Jacomo
- & Leon Danon
-
Article
| Open Access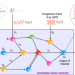 Frequency spectra and the color of cellular noise
Frequency spectra and the color of cellular noise
The invention of the Fourier integral in the 19th century laid the foundation for modern spectral analysis methods. Here the authors develop frequency-based methods for analyzing the reaction mechanisms within living cells from distinctively noisy single-cell output trajectories and present forward engineering of synthetic oscillators and controllers.
- Ankit Gupta
- & Mustafa Khammash
-
Article
| Open Access Discovering the drivers of clonal hematopoiesis
Discovering the drivers of clonal hematopoiesis
Identifying the genetic drivers of clonal haematopoiesis (CH) has been challenging due to their low frequencies and a lack of adequate tools. Here, the authors use a reverse calling to detect blood somatic mutations and the IntOGen pipeline to identify CH drivers in large cancer genomics data sets based on signals of positive selection.
- Oriol Pich
- , Iker Reyes-Salazar
- & Nuria Lopez-Bigas
-
Article
| Open Access Accurate somatic variant detection using weakly supervised deep learning
Accurate somatic variant detection using weakly supervised deep learning
Deep learning could be applied to the challenge of somatic variant calling in cancer by making use of large-scale genomic data. Here, the authors develop VarNet, a weakly supervised deep learning model for somatic variant calling in cancer with robust performance across multiple cancer genomics datasets.
- Kiran Krishnamachari
- , Dylan Lu
- & Anders Jacobsen Skanderup
-
Article
| Open Access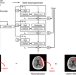 Emergency triage of brain computed tomography via anomaly detection with a deep generative model
Emergency triage of brain computed tomography via anomaly detection with a deep generative model
Triage is essential for the early diagnosis and reporting of emergency patients in the emergency department. Here, the authors develop an anomaly detection algorithm with a deep generative model that reprioritizes radiology worklists and provides lesion attention maps for brain CT images with critical findings.
- Seungjun Lee
- , Boryeong Jeong
- & Namkug Kim
-
Article
| Open Access Myasthenia gravis-specific aberrant neuromuscular gene expression by medullary thymic epithelial cells in thymoma
Myasthenia gravis-specific aberrant neuromuscular gene expression by medullary thymic epithelial cells in thymoma
Myasthenia Gravis and thymoma are frequently associated with patients suffering from both diseases. Here the authors perform single cell sequencing of thymoma and find that there are autoimmune antigens such as neuromuscular proteins expressed aberrantly in neuromuscular mTECs in patients with both diseases.
- Yoshiaki Yasumizu
- , Naganari Ohkura
- & Hideki Mochizuki
-
Article
| Open Access A multi-omic dissection of super-enhancer driven oncogenic gene expression programs in ovarian cancer
A multi-omic dissection of super-enhancer driven oncogenic gene expression programs in ovarian cancer
Super-enhancers and their associated transcription factor networks have been shown to influence ovarian cancer biology. Here, based on an integrated set of genomic and epigenomic datasets, the authors identify clinically relevant super-enhancers amplified in ovarian cancer patients and functionally validate their activity.
- Michael R. Kelly
- , Kamila Wisniewska
- & Hector L. Franco
-
Article
| Open Access Differential analysis of RNA structure probing experiments at nucleotide resolution: uncovering regulatory functions of RNA structure
Differential analysis of RNA structure probing experiments at nucleotide resolution: uncovering regulatory functions of RNA structure
The authors present DiffScan, an advanced tool for normalization and differential analysis of RNA structure probing experiments, combining their power in deciphering the dynamic RNA structurome and facilitating the discovery of RNA regulatory functions.
- Bo Yu
- , Pan Li
- & Lin Hou
-
Article
| Open Access A Bayesian approach to infer recombination patterns in coronaviruses
A Bayesian approach to infer recombination patterns in coronaviruses
Genetic recombination can confound standard phylogenetic approaches. Here, the authors present a method to reconstruct virus recombination networks, and show the importance of recombination in shaping the ongoing evolution of SARS-like, MERS and 3 human seasonal coronaviruses.
- Nicola F. Müller
- , Kathryn E. Kistler
- & Trevor Bedford
-
Article
| Open Access Drivers and trends of global soil microbial carbon over two decades
Drivers and trends of global soil microbial carbon over two decades
Soil microbial carbon is central to soil functions and services, but its spatial-temporal dynamics are unclear. Here the authors show global trends in soil microbial carbon, which suggests a global decrease in soil microbial carbon, mostly driven by temperature increases in northern areas.
- Guillaume Patoine
- , Nico Eisenhauer
- & Carlos A. Guerra
-
Article
| Open Access People infer communicative action through an expectation for efficient communication
People infer communicative action through an expectation for efficient communication
Humans can quickly infer when someone’s body movements are meant to be communicative. Here, the authors show that this capacity is underpinned by an expectation that communicative actions will efficiently reveal that they lack an external goal.
- Amanda Royka
- , Annie Chen
- & Julian Jara-Ettinger
-
Article
| Open Access Identifying multicellular spatiotemporal organization of cells with SpaceFlow
Identifying multicellular spatiotemporal organization of cells with SpaceFlow
A critical task in spatial transcriptomics analysis is to understand inherently spatial relationships among cells. Here, the authors present a deep learning framework to integrate spatial and transcriptional information, spatially extending pseudotime and revealing spatiotemporal organization of cells.
- Honglei Ren
- , Benjamin L. Walker
- & Qing Nie
-
Article
| Open Access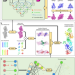 Model building of protein complexes from intermediate-resolution cryo-EM maps with deep learning-guided automatic assembly
Model building of protein complexes from intermediate-resolution cryo-EM maps with deep learning-guided automatic assembly
One challenge in cryo-EM is to build atomic models into intermediate resolution maps. Here, the authors present a deep learning-guided iterative assembling method by integrating AlphaFold, FFTbased fitting, and domain-based refinement.
- Jiahua He
- , Peicong Lin
- & Sheng-You Huang
-
Comment
| Open AccessCommunity voices: policy proposals to promote inclusion in academia through the lens of women in science
Diversity is a creative force that broadens views and enhances ideas; it increases productivity as well as the impact of our science, making our respective organisations more agile and timely. Equality of opportunity is a key to success for any research organisation. Here we argue that every research organisation, whether in academia or in industry, needs to have better inclusion policies to harness the benefits of diversity in research. Drawing from our personal experiences and perspectives as women in science, we share our suggestions on how to promote inclusion in academia and create a better research culture for all. Our shared experiences highlight the many hurdles women in science face on a daily basis. We stress that rules and regulations, as well as education for awareness, will play critical role in this much needed shift from a male-dominated scientific culture that dates from Victorian times to a modern focus on gender equality in science. The key ingredients of this new culture will be flexibility, transparency, fairness and thoughtfulness.
- Sarah A. Teichmann
- , Muzlifah Haniffa
- & Jasmin Fisher
-
Article
| Open Access A reference single-cell regulomic and transcriptomic map of cynomolgus monkeys
A reference single-cell regulomic and transcriptomic map of cynomolgus monkeys
Non-human primates are attractive laboratory animal models that can accurately reflect some developmental and pathological features of humans. Here the authors chart a reference cell map of cynomolgus monkeys using both scATAC-seq and scRNA-seq data across multiple organs, providing insights into the molecular dynamics and cellular heterogeneity of this organism.
- Jiao Qu
- , Fa Yang
- & Dijun Chen
-
Article
| Open Access Integration of tumor extrinsic and intrinsic features associates with immunotherapy response in non-small cell lung cancer
Integration of tumor extrinsic and intrinsic features associates with immunotherapy response in non-small cell lung cancer
Some cancer patients with impaired HLA-I still respond to immunotherapy. Here the authors combine a cytotoxic gene signature from CD4+ and CD8+ T cells with tumor mutational burden to predict immunotherapy response in NSCLC patients, including those with HLA-LOH.
- Denise Lau
- , Sonal Khare
- & Aly A. Khan
-
Article
| Open Access Deciphering polymorphism in 61,157 Escherichia coli genomes via epistatic sequence landscapes
Deciphering polymorphism in 61,157 Escherichia coli genomes via epistatic sequence landscapes
Predicting the effects of mutations in a species is a major challenge in genetics. Here, the authors investigate protein sequence landscapes using diverged E. coli sequences, to predict tolerated mutations and capture interactions between mutations.
- Lucile Vigué
- , Giancarlo Croce
- & Martin Weigt
-
Article
| Open Access Single-cell analysis highlights differences in druggable pathways underlying adaptive or fibrotic kidney regeneration
Single-cell analysis highlights differences in druggable pathways underlying adaptive or fibrotic kidney regeneration
After acute injury, kidneys either successfully repair/regenerate or become fibrotic. Here the authors use scRNA-seq to study adaptive/maladaptive kidney regeneration and identify proinflammatory/fibrotic proximal tubule cells with pharmacologically targetable pyroptosis/ferroptosis signatures.
- Michael S. Balzer
- , Tomohito Doke
- & Katalin Susztak
-
Article
| Open Access Machine learning-based tissue of origin classification for cancer of unknown primary diagnostics using genome-wide mutation features
Machine learning-based tissue of origin classification for cancer of unknown primary diagnostics using genome-wide mutation features
The original tumor location can be unclear for metastatic tumors. Here, the authors show that DNA sequencing of whole genomes can be used to classify metastatic tumors using a machine learning model, Cancer of Unknown Primary Location Resolver, in order to improve diagnosis and inform treatment decisions.
- Luan Nguyen
- , Arne Van Hoeck
- & Edwin Cuppen
-
Article
| Open Access Recurrent somatic mutations as predictors of immunotherapy response
Recurrent somatic mutations as predictors of immunotherapy response
Few genetic biomarkers are known for cancer immunotherapy. Here the authors identify recurrently-mutated genes and pathways associated with treatment response and develop a classifier using tumour whole exome sequencing and clinical features.
- Zoran Z. Gajic
- , Aditya Deshpande
- & Neville E. Sanjana
-
Article
| Open Access Mimicked synthetic ribosomal protein complex for benchmarking crosslinking mass spectrometry workflows
Mimicked synthetic ribosomal protein complex for benchmarking crosslinking mass spectrometry workflows
Cross-linking mass spectrometry is widely used to elucidate protein structures and interactions. Here, the authors generate an extensive peptide library to benchmark the most common cross-link search engines with frequently used cross-linking reagents in low and high complex sample systems.
- Manuel Matzinger
- , Adrian Vasiu
- & Karl Mechtler
-
Article
| Open Access Deep learning from phylogenies to uncover the epidemiological dynamics of outbreaks
Deep learning from phylogenies to uncover the epidemiological dynamics of outbreaks
Widely applicable, accurate and fast inference methods in phylodynamics are needed to fully profit from the richness of genetic data in uncovering the dynamics of epidemics. Here, the authors develop a likelihood-free, simulation-based deep learning approach.
- J. Voznica
- , A. Zhukova
- & O. Gascuel
-
Article
| Open Access Loss-of-function, gain-of-function and dominant-negative mutations have profoundly different effects on protein structure
Loss-of-function, gain-of-function and dominant-negative mutations have profoundly different effects on protein structure
Most known pathogenic mutations occur in protein-coding regions of DNA and change the way proteins are made. Here the authors analyse the locations of thousands of human disease mutations and their predicted effects on protein structure and show that,while loss-of-function mutations tend to be highly disruptive, non-loss-of-function mutations are in general much milder at a protein structural level.
- Lukas Gerasimavicius
- , Benjamin J. Livesey
- & Joseph A. Marsh
-
Article
| Open Access A versatile active learning workflow for optimization of genetic and metabolic networks
A versatile active learning workflow for optimization of genetic and metabolic networks
Optimization of biological networks is often limited by wet lab labor and cost, and the lack of convenient computational tools. Here, aimed at democratization and standardization, the authors describe METIS, a modular and versatile active machine learning workflow with a simple online interface for the optimization of biological target functions with minimal experimental datasets.
- Amir Pandi
- , Christoph Diehl
- & Tobias J. Erb
-
Article
| Open Access Self-evolving vision transformer for chest X-ray diagnosis through knowledge distillation
Self-evolving vision transformer for chest X-ray diagnosis through knowledge distillation
Although deep learning-based computer-aided diagnosis systems have recently achieved expert level performance, developing a robust model requires large, high-quality data with annotations. Here, the authors present a framework which can improve the performance of vision transformer simultaneously with self-supervision and self-training.
- Sangjoon Park
- , Gwanghyun Kim
- & Jong Chul Ye
-
Article
| Open Access A convolutional neural network highlights mutations relevant to antimicrobial resistance in Mycobacterium tuberculosis
A convolutional neural network highlights mutations relevant to antimicrobial resistance in Mycobacterium tuberculosis
Pathogen whole genome sequencing, coupled with statistical and machine learning models, offers a promising solution to multi-drug resistance diagnosis. Here, the authors present two deep convolutional neural networks that predict the antibiotic resistance phenotypes of M. tuberculosis isolates.
- Anna G. Green
- , Chang Ho Yoon
- & Maha Farhat
-
Article
| Open Access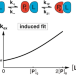 A litmus test for classifying recognition mechanisms of transiently binding proteins
A litmus test for classifying recognition mechanisms of transiently binding proteins
The authors provide a litmus test for the recognition mechanism of transiently binding proteins based on nuclear magnetic resonance and find a conformational selection binding mechanism through concentration-dependent kinetics of ubiquitin and SH3.
- Kalyan S. Chakrabarti
- , Simon Olsson
- & Christian Griesinger
-
Article
| Open Access Co-optimization of therapeutic antibody affinity and specificity using machine learning models that generalize to novel mutational space
Co-optimization of therapeutic antibody affinity and specificity using machine learning models that generalize to novel mutational space
Optimising antibody properties such as affinity can be detrimental to other key properties. Here the authors use machine learning to simplify the identification of antibodies with co-optimal levels of affinity and specificity for a clinical-stage antibody that displays high levels of on- and off-target binding.
- Emily K. Makowski
- , Patrick C. Kinnunen
- & Peter M. Tessier
-
Article
| Open Access Many dissimilar NusG protein domains switch between α-helix and β-sheet folds
Many dissimilar NusG protein domains switch between α-helix and β-sheet folds
Folded proteins are composed of secondary structures, α-helices and β-sheets, that are generally assumed to be stable. Here, the authors combine computational prediction with experimental validation to show that many sequence-diverse NusG protein domains switch completely from α-helix to β-sheet folds.
- Lauren L. Porter
- , Allen K. Kim
- & Marie-Paule Strub
-
Article
| Open Access Reconstruction of a catalogue of genome-scale metabolic models with enzymatic constraints using GECKO 2.0
Reconstruction of a catalogue of genome-scale metabolic models with enzymatic constraints using GECKO 2.0
Genome-scale metabolic models have been widely used for quantitative exploration of the relation between genotype and phenotype. Here the authors present GECKO 2, an automated framework for continuous and version controlled update of enzyme-constrained models of metabolism, producing an interesting catalogue of high-quality models for diverse yeasts, bacteria and human metabolism, aiming to facilitate their use in basic science, metabolic engineering and synthetic biology purposes.
- Iván Domenzain
- , Benjamín Sánchez
- & Jens Nielsen
-
Article
| Open Access Pervasive translation of circular RNAs driven by short IRES-like elements
Pervasive translation of circular RNAs driven by short IRES-like elements
Unbiased screen of random sequences identified many short IRES-like elements to drive circular RNA translation and hundreds of rolling circle translation events, suggesting a pervasive cap-independent translation in human transcriptome.
- Xiaojuan Fan
- , Yun Yang
- & Zefeng Wang
-
Article
| Open Access Vertex protein PduN tunes encapsulated pathway performance by dictating bacterial metabolosome morphology
Vertex protein PduN tunes encapsulated pathway performance by dictating bacterial metabolosome morphology
Morphology of metabolosomes affects the encapsulated pathway performance. Here, the authors combine experimental characterizations with structural and kinetic modeling to reveal how the shell protein PduN changes the morphology of 1,2-propanediol utilization (Pdu) metabolosome and how this morphology shift impacts Pdu function.
- Carolyn E. Mills
- , Curt Waltmann
- & Danielle Tullman-Ercek
-
Article
| Open Access Deep learning to diagnose Hashimoto’s thyroiditis from sonographic images
Deep learning to diagnose Hashimoto’s thyroiditis from sonographic images
Hashimoto’s thyroiditis (HT) is the main cause of hypothyroidism. Here the authors develop a deep learning model for diagnosis of HT on a large multi-site dataset including image and video data.
- Qiang Zhang
- , Sheng Zhang
- & Xiangchun Li
-
Article
| Open Access The impact of rare germline variants on human somatic mutation processes
The impact of rare germline variants on human somatic mutation processes
The impact of germline variants on somatic alterations in cancer remains to be explored in large-scale datasets. Here, the authors study the association of rare germline variants with somatic mutational processes in more than 15,000 tumors, and reveal that damaging variants in newly-identifed genes are prevalent in the population.
- Mischan Vali-Pour
- , Solip Park
- & Fran Supek
-
Article
| Open Access Learning representations of chromatin contacts using a recurrent neural network identifies genomic drivers of conformation
Learning representations of chromatin contacts using a recurrent neural network identifies genomic drivers of conformation
Despite the availability of chromatin conformation capture experiments, discerning the relationship between the 1D genome and 3D conformation remains a challenge. Here, the authors propose a method that produces low-dimensional latent representations that summarize intra-chromosomal Hi-C contacts.
- Kevin B. Dsouza
- , Alexandra Maslova
- & Maxwell W. Libbrecht
-
Article
| Open Access Clonal reconstruction from co-occurrence of vector integration sites accurately quantifies expanding clones in vivo
Clonal reconstruction from co-occurrence of vector integration sites accurately quantifies expanding clones in vivo
High transduction rates of viral vectors ensure good gene delivery; however multiple integration events can occur in the same cell. Here the authors use correlations between repeated measurements of integration site abundances to estimate their mutual similarity and identify clusters of co-occurring sites.
- Sebastian Wagner
- , Christoph Baldow
- & Ingmar Glauche
-
Article
| Open Access Network-based machine learning approach to predict immunotherapy response in cancer patients
Network-based machine learning approach to predict immunotherapy response in cancer patients
Identifying biomarkers for response to immunotherapy in cancer remains challenging. Here, the authors develop an approach based on network biology and machine learning -NetBio- to identify molecular biomarkers of response to immunotherapy across different cancer types and cohorts.
- JungHo Kong
- , Doyeon Ha
- & Sanguk Kim
-
Article
| Open Access Context-aware deconvolution of cell–cell communication with Tensor-cell2cell
Context-aware deconvolution of cell–cell communication with Tensor-cell2cell
Cellular contexts such as disease state, organismal life stage and tissue microenvironment, shape intercellular communication, and ultimately affect an organism’s phenotypes. Here, the authors present Tensor-cell2cell, an unsupervised method for deciphering context-driven intercellular communication.
- Erick Armingol
- , Hratch M. Baghdassarian
- & Nathan E. Lewis
-
Article
| Open Access GenomicSuperSignature facilitates interpretation of RNA-seq experiments through robust, efficient comparison to public databases
GenomicSuperSignature facilitates interpretation of RNA-seq experiments through robust, efficient comparison to public databases
Many transcriptomic profiles have been deposited in public archives but are underused for the interpretation of experiments. Here the authors report GenomicSuperSignature for interpreting new transcriptomic datasets through comparison to public archives, without high-performance computing requirements.
- Sehyun Oh
- , Ludwig Geistlinger
- & Sean Davis
-
Article
| Open Access Occult polyclonality of preclinical pancreatic cancer models drives in vitro evolution
Occult polyclonality of preclinical pancreatic cancer models drives in vitro evolution
It is unclear if the molecular profiles of pancreatic ductal adenocarcinoma (PDAC) preclinical models remain stable during propagation. Here, the authors characterise clonal evolution throughout propagation in PDAC cell lines and a patient-derived organoid using single-cell genomics, transcriptomics and epigenomics.
- Maria E. Monberg
- , Heather Geiger
- & Anirban Maitra
-
Article
| Open Access A microfluidic optimal experimental design platform for forward design of cell-free genetic networks
A microfluidic optimal experimental design platform for forward design of cell-free genetic networks
Characterization of cell-free genetic networks is inherently difficult. Here the authors use optimal experimental design and microfluidics to improve characterization, demonstrating modularity and predictability of parts in applied test cases.
- Bob van Sluijs
- , Roel J. M. Maas
- & Wilhelm T. S. Huck
-
Article
| Open Access Contribution of low population immunity to the severe Omicron BA.2 outbreak in Hong Kong
Contribution of low population immunity to the severe Omicron BA.2 outbreak in Hong Kong
Hong Kong experienced a severe wave of SARS-CoV-2 in early 2022. Here, the authors use genomic and serosurveillance data and show that this wave was dominated by the Omicron BA.2 sublineage, and that low protective immunity, particularly in older age groups, contributed to its severity.
- Lin-Lei Chen
- , Syed Muhammad Umer Abdullah
- & Kelvin Kai-Wang To
-
Article
| Open Access Endothelial cell heterogeneity and microglia regulons revealed by a pig cell landscape at single-cell level
Endothelial cell heterogeneity and microglia regulons revealed by a pig cell landscape at single-cell level
Pigs are important large animal models for biomedical research. Here, the authors construct a single-cell landscape of pig tissues, unravelling the phenotypic heterogeneity of blood endothelial cells in adipose tissues and the evolutionally conserved regulons of microglia in brains.
- Fei Wang
- , Peiwen Ding
- & Yonglun Luo
-
Article
| Open Access Probing TDP-43 condensation using an in silico designed aptamer
Probing TDP-43 condensation using an in silico designed aptamer
Here, the authors generated an artificial RNA molecule, or aptamer, specific for the Amyotrophic Lateral Sclerosis protein TDP-43. By interacting avidly with its target, the aptamer can be exploited to track TDP-43 phase transition in vitro and in cells.
- Elsa Zacco
- , Owen Kantelberg
- & Gian Gaetano Tartaglia
-
Article
| Open Access Forest Fire Clustering for single-cell sequencing combines iterative label propagation with parallelized Monte Carlo simulations
Forest Fire Clustering for single-cell sequencing combines iterative label propagation with parallelized Monte Carlo simulations
In the era of single-cell sequencing, there is a growing need to extract insights from data with clustering methods. Here, inspired by forest fire dynamics, the authors devise an algorithm that can cluster single-cell data with minimal prior assumptions and can compute a non-parametric posterior probability for each data point.
- Zhanlin Chen
- , Jeremy Goldwasser
- & Mark Gerstein
-
Article
| Open Access HarmonizR enables data harmonization across independent proteomic datasets with appropriate handling of missing values
HarmonizR enables data harmonization across independent proteomic datasets with appropriate handling of missing values
Dataset integration is common practice to overcome limitations in statistically underpowered omics datasets. Here the authors present “HarmonizR”, a tool for missing data tolerant experimental variance reduction in large, integrated but independently generated datasets without data imputation, adjustable for individual dataset modalities, correction algorithm, and user preferences.
- Hannah Voß
- , Simon Schlumbohm
- & Christoph Krisp
-
Article
| Open Access Global stable-isotope tracing metabolomics reveals system-wide metabolic alternations in aging Drosophila
Global stable-isotope tracing metabolomics reveals system-wide metabolic alternations in aging Drosophila
Stable-isotope tracing allows quantifying metabolic activity by measuring isotopically labeled metabolites, but its metabolome coverage has been limited. Here, the authors develop a global isotope tracing approach with metabolome-wide coverage and use it to characterize metabolic activities in aging Drosophila.
- Ruohong Wang
- , Yandong Yin
- & Zheng-Jiang Zhu
 Aggregative trans-eQTL analysis detects trait-specific target gene sets in whole blood
Aggregative trans-eQTL analysis detects trait-specific target gene sets in whole bloodBrowse broader subjects
Browse narrower subjects
- Biochemical reaction networks
- Cellular signalling networks
- Classification and taxonomy
- Communication and replication
- Computational models
- Computational neuroscience
- Computational platforms and environments
- Data acquisition
- Data integration
- Data mining
- Data processing
- Data publication and archiving
- Databases
- Functional clustering
- Gene ontology
- Gene regulatory networks
- Genome informatics
- Hardware and infrastructure
- High-throughput screening
- Image processing
- Literature mining
- Machine learning
- Microarrays
- Network topology
- Phylogeny
- Power law
- Predictive medicine
- Probabilistic data networks
- Programming language
- Protein analysis
- Protein design
- Protein folding
- Protein function predictions
- Protein structure predictions
- Proteome informatics
- Quality control
- Scale invariance
- Sequence annotation
- Software
- Standards
- Statistical methods
- Virtual drug screening
