


 Figure 1: Adenovirus E1A stabilizes Myc
Figure 1: Adenovirus E1A stabilizes Myc















« Prev Next »
Although some human diseases, such as cystic fibrosis, are caused by mutations in a single gene, the majority of disorders, including heart disease, cancer, and bipolar disorder, are the result of multiple causes. In these and other so-called multifactorial diseases, multiple gene products and environmental factors interact to influence disease etiology in complex ways. Knowledge of how gene products interact at the protein level is thus important for understanding how diseases develop — as well as how they can be stopped.
Insight into the molecular basis of complex diseases — specifically, cancers — has been provided by multiple studies examining the interaction between oncogenes (tumor-promoting genes) and tumor suppressor genes during cancer progression. This article takes a closer look at several such studies, examining various examples of how environmental factors, such as viral proteins, can interact with normal proteins to lead to cancer.
Before considering the aforementioned studies in greater depth, let's take a brief moment to review the process by which viruses multiply. Recall that after infecting a host cell, viruses hijack the cellular machinery to replicate their DNA. This is often accompanied by the induction of cell division in the host cell, because DNA replication is a key component of the cell cycle. In this way, some viruses are able to promote cellular transformation, which causes cancer because these viruses are able to encourage a cell to divide when it normally would not. Indeed, viruses often are able to lift the restriction that most cells have on cell division by changing the expression pattern of proteins that control progression through the cell cycle, including the proteins produced by tumor suppressor genes and oncogenes.
Viral Proteins Can Directly Interfere with Tumor Suppressors
The discovery that viral proteins bind to and inhibit normal cellular proteins, such as cell cycle regulators, revealed one possible mechanism by which environmental factors (in this case, viruses) could cause cancer. For instance, proteins made by several oncogenic (cancer-causing) viruses, including adenovirus, SV40, and human papillomavirus, were found to bind to the tumor suppressor protein called retinoblastoma (Rb) (Whyte et al., 1988; DeCaprio et al., 1988; Dyson et al., 1989). Rb plays an important role in regulating cell proliferation by keeping the cell cycle in check. Interfering with Rb can lead to unregulated proliferation; therefore, not surprisingly, mutations in the Rb gene can lead to cancers in many different cell types. The observation that viral oncoproteins can physically bind Rb, however, provided a unique opportunity to learn about how viruses and viral proteins can cause cancer. In fact, insight into the molecular mechanisms underlying viral oncogenicity came from examining the consequences of viral oncoprotein binding to Rb.
Normally, the cellular transcription factor E2F promotes cell division by facilitating progression through the S phase of mitosis (Giacinti & Giordano, 2006). E2F activity is typically inhibited by Rb, which prevents cell division. However, when the adenovirus E1A protein is expressed in cells after infection, this protein competes with E2F to bind to Rb (Dyson et al., 1992; Bagchi et al., 1990). When Rb is pulled away from E2F, it can no longer inhibit E2F from activating the transcription of genes that up-regulate cell division. Thus, E2F's target genes are transcribed, and the cell cycle proceeds unchecked. These observations help explain how an environmental agent, such as a virus, actually interferes with normal regulation of gene expression. In this case, changes in gene expression promote cell division and cellular transformation, which leads to cancer.
E1A Alters Expression of a Key Tumor Suppressor Protein
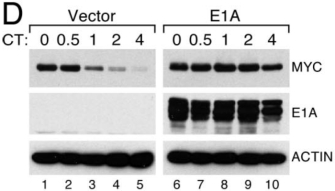
© 2008 National Academy of Sciences, USA Tworkowski, K. A. et al. Adenovirus E1A targets p400 to induce the cellular oncoprotein Myc. PNAS 105, 6103-6108. All rights reserved. 
Gene expression is not only regulated at the "front end," or at the level of transcription, however. After a gene is expressed as a protein, that protein has a certain half-life. The longer the protein is around, the longer it can continue to do its job. Thus, the rate at which proteins are degraded in cells is highly regulated, and for good reason. For example, in other studies examining adenoviruses (also called DNA tumor viruses), investigators showed that cellular transformation was facilitated by stabilizing specific proteins that might otherwise be degraded. One such protein was E1A. In addition to binding to Rb, E1A has been shown to interact with other cellular proteins, including a complex that includes the protein p400. Indeed, the interaction between E1A and p400 is so important that a mutant of E1A that can no longer interact with p400 cannot cause cellular transformation (Fuchs et al., 2001).
It was already known that E1A expression resulted in the stabilization of an oncogene called Myc. Specifically, when levels of Myc protein were followed over time in cells also expressing E1A, it was shown that Myc protein was present longer in those cells than their non-E1A-expressing counterparts (Figure 1). It was thus hypothesized that the stabilization occurred when E1A interfered with the degradation of Myc by proteosomes, one method that cells use to control protein turnover. However, Tworkowski et al. (2008) found that the viral protein E1A actually enabled Myc protein to form a complex with p400 that was resistant to degradation. When it was not degraded, the Myc then acted as a transcription factor, turning on the expression of downstream target genes that promoted cell division and ultimately cellular transformation.
Studies of the interactions between tumor suppressor genes, oncogenes, and environmental factors such as viruses show how these interactions influence gene expression and have greatly increased our understanding of why disease is so complex. These studies indicate that the molecular basis of complex disease is not always a result of genetic mutation, which is often a focus of research. Instead, changes in the expression of a wild-type gene caused by external influences can often have profound developmental and pathological implications. Knowledge of the molecular mechanisms underlying this complexity has provided valuable information about normal cellular processes and why they sometimes go wrong. Moreover, these studies have provided exciting possibilities for the design and execution of therapies aimed at either restoring the activity of tumor suppressor genes or interfering with oncogenes to alter cancer progression.
References and Recommended Reading
Bagchi, S., et al. Adenovirus E1A proteins can dissociate heteromeric complexes involving the E2F transcription factor: A novel mechanism for E1A trans-activation. Cell 62, 659–669 (1990)
Baker, S. J., et al. Suppression of human colorectal carcinoma cell growth by wild-type p53. Science 249, 912–915 (1990)
DeCaprio, J. A., et al. SV40 large tumor antigen forms a specific complex with the product of the retinoblastoma susceptibility gene. Cell 54, 275–283 (1988)
Dyson, N., et al. The human papilloma virus-16 E7 oncoprotein is able to bind to the retinoblastoma gene product. Science 243, 934–937 (1989)
Dyson, N., et al. Homologous sequences in adenovirus E1A and human papillomavirus E7 proteins mediate interaction with the same set of cellular proteins. Journal of Virology 66, 6893–6902 (1992)
Fuchs, M., et al. The p400 complex is an essential E1A transformation target. Cell 106, 297–307 (2001)
Giancinti, C., & Giordano, A. RB and cell cycle progression. Oncogene 25, 5220–5227 (2006)
Ogawa, H., et al. A complex with chromatin modifiers that occupies E2F- and Myc-responsive genes in G0 cells. Science 296, 1132–1136 (2002)
Tworkowski, K., et al. Adenovirus E1A targets p400 to induce the cellular oncoprotein Myc. Proceedings of the National Academy of Sciences 105, 6103–6108 (2008)
Whyte, P., et al. Association between an oncogene and an anti-oncogene: The adenovirus E1A proteins bind to the retinoblastoma gene product. Nature 334, 124–129 (1988) (link to article)









