


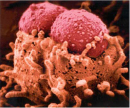
 Figure 2
Figure 2
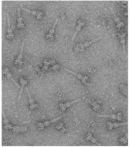















« Prev Next »
Worldwide, infectious diseases account for more than 10 million deaths each year (World Health Organization, 2004). In the past decade, exciting developments in molecular genetics have allowed scientists to explore the complex pathways used by microorganisms to elicit these diseases. So, what do we know so far?
Back in the time of Hippocrates, when microorganisms were virtually unimagined, doctors believed that disease stemmed from an imbalance in the four bodily fluids: blood, phlegm, yellow bile, and black bile. Then, in the 1670s, Dutch cloth merchant-turned-microbiologist Antonie van Leeuwenhoek offered the first observations of "animalicules" swimming in droplets of rainwater magnified under his microscope. But it wasn't until the late 1800s that early microbiologists began putting two and two together, eventually determining that microorganisms cause disease. But how does this happen?
Common Themes in the Genetics of Virulence
Unlocking the genetic code of disease-causing microorganisms has helped identify common features associated with their virulence. These include pathogenicity islands, the incorporation of bacteriophage genes, and abnormal amounts of guanine and cytosine in the sequences of virulence genes.
Pathogenicity Islands
As the genomic sequences of viruses, fungi, and bacteria become increasingly available, scientists have noticed that the genes involved in disease often appear as groups. In bacteria, these groups are called pathogenicity islands, and they can be part of a chromosome or found on separate plasmids. These islands often contain genes required for the pathogen to enter a host, or genes that promote the pathogen's survival once it gains entry.
Consider, for example, the pathogenicity islands encoded in the chromosome of Escherichia coli O157:H7 (Figure 1). This enterohemorrhagic bacterium was implicated in a 2006 raw spinach outbreak that sickened hundreds of people across the United States and killed three of them. The red and tan boxes on the outer ring of Figure 1 indicate clusters of genes—or islands—that are found in the virulent strain of E. coli but not in the nonpathogenic laboratory strain, E. coli K12 (Perna, 2001). Nearly 30 percent of the 5,416 genes encoded by pathogenic E. coli lie within these islands. Some are putative virulence factors, meaning that they have been recently identified and warrant further investigation. Others are well-established players in the disease process.
But just how might these clusters help E. coli cause disease? To answer this question, start by examining the LEE pathogenicity island, which is found just before the origin of replication in Figure 1. LEE stands for "locus of enterocyte effacement," and this island is an excellent example of a gene cluster that is rich in virulence factors (Elliott, 1998). The genes encoded in this island allow enterohemorrhagic E. coli to attach to the intestinal mucosa and secrete toxins that cause bloody diarrhea, or hemorrhagic colitis—a hallmark of infection with this organism. One of the more notorious genes on this island is eae, which stands for "E. coli attaching and effacing." The eae gene expresses intimin, an outer membrane surface adhesin that induces the formation of attaching and effacing lesions—intestinal mucosal changes that were originally observed in the intestines of animals that had been infected with enteropathogenic E. coli (Moon et al., 1983). Intestinal epithelial cells respond to these lesions by forming convenient actin pedestals for the bacteria to sit on (Figure 2). Intimin does not function alone, however; before it can take effect, several other genes on the LEE island must be expressed. In particular, the sep genes must collaborate to build a type III secretion system; this is a molecular "syringe" that is a relatively common virulence factor expressed by many pathogens to inject toxins into host cells (Figure 3). One of the molecules injected by the E. coli type III secretion system is the translocated intimin receptor, or Tir. Intimin then binds to Tir and induces formation of the lesions. This molecular system is just one example of the many diverse virulence mechanisms encoded in the bacterial genome.
Bacteriophages
Returning to the pathogenicity islands depicted in Figure 1, note that there is a red box labeled BP-933W found just above the terminus of replication. This box corresponds to a bacteriophage, or a bacteria-infecting virus. Bacteriophages often integrate into microbial chromosomes and encode genes involved in virulence. Because most bacteriophages are capable of lytic replication, these viruses can hop out of the chromosome—in response to DNA damage, for example—and infect other organisms, conferring virulence traits. For example, in E. coli, the BP-933W bacteriophage encodes Shiga toxin 2 (Stx2), a substance that enters and kills host cells. Along with an additional Shiga toxin known as Stx1 (which is encoded elsewhere on the chromosome), Stx2 plays a key role in the development of hemorrhagic colitis and hemolytic uremic syndrome, the number-one cause of acute renal failure among children.
As is the case for many virulence factors, mutation of the stx2 gene doesn't affect a bacterium's ability to grow (Figure 4a) or to produce other disease-related proteins, such as intimin or Tir (Figure 4b); however, it does limit the bacterium's ability to harm host cells. As shown in Figure 4c, the stx2 mutant is no more cytotoxic than growth medium in Vero cells, which were derived from the kidney epithelial cells of African green monkeys and are commonly used in studies of bacterial virulence factors.
Guanine and Cytosine Content
Having considered some of the virulence factors encoded by E. coli, take another look at Figure 1. The second-largest circle illustrates another important concept in molecular genetics—specifically, the sequences of virulence genes often contain different amounts of guanine and cytosine (i.e., G + C) residues than are found in the rest of the genome. For instance, in the smaller circle beneath the red and tan blocks, notice the peaks in G + C content as compared with the surrounding regions.
The genomic G + C content ranges from 25% to 75% among eubacteria. This variation likely reflects the recent acquisition of these genes from a foreign source through horizontal gene transfer. This makes a lot of sense when considering that pathogenicity islands often begin and end with insertion sequences that allow the virulence genes to hop from loci to loci, or from organism to organism. These insertion sequences are sometimes flanked by inverted repeats, or palindromes, which are thought to result when DNA from another organism is inserted into a chromosome. Insertion sequences may explain how certain strains, like E. coli O157:H7, evolved to become pathogenic in the first place, while other strains of the same species, like E. coli K12, typically do not cause disease.
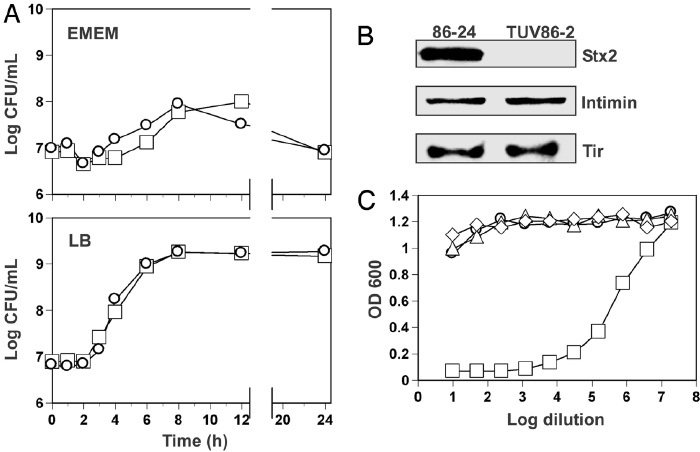
Figure 4: Growth comparison, expression profiles, and characterization of cytotoxicity associated with WT EHEC and its stx2 isogenic mutant.
(A) Growth curves of wild-type E. coli O157:H7 (squares) and an isogenic mutant of stx2 (circles) in Eagle's minimal essential medium (EMEM) and LB, a medium that is relatively rich in nutrients. (B) Immunoblots of cell lysates from the wild-type (i.e., 86-24) and mutant (i.e., TUV86-2) strains, showing expression of the Stx2, Intimin, and Tir proteins. (C) Cytotoxicity of the stx2 mutant (circle), growth medium (diamond), nonpathogenic E. coli DH5a (i.e., a laboratory strain commonly used for genetic cloning; triangle), and the wild-type strain (square) in Vero cells.
© 2006 National Academy of Sciences, USA Robinson, C. M., et al. Shiga toxin of enterohemorrhagic Escherichia coli type O157:H7 promotes intestinal colonization. Proceedings of the National Academy of Sciences 103, 9667–9672 (2006) doi:10.1073/pnas.0602359103. All rights reserved. 
Exceptions to the Rule
Of course, in such discussions, it is important to note that "typically" is the operative word. In fact, normally avirulent strains of E. coli can—and do—cause disease when they wander into the wrong places at the wrong times. For example, many women have experienced the painful burn of a urinary tract infection when commensal E. coli from the digestive tract takes a wrong turn up the urethra. Such phenomena raise another important point: Identifying the virulence genes encoded by notorious pathogens (like E. coli O157:H7) has been only part of the microscopic puzzle. Over the years, researchers increasingly began to notice that some organisms cause disease only when they appear at certain sites at certain times, whereas others cause trouble no matter where and when they turn up. Thus, the other major part of the puzzle involved explaining and predicting why this happens.
Consider this: Candida albicans generally colonizes the skin and mucous membranes without much difficulty. However, this fungus can cause vaginitis, nail infections, and even toxic shock if the mucosal barrier—of the genital tract, for example—is disrupted, the immune system is suppressed by drug therapy, or someone is exposed to an especially large inoculum. In fact, in 1969, German physician Wolfgang von Krause imbibed a concoction of approximately 1 billion C. albicans to prove that the normally docile fungus can indeed cause disease. Unfortunately, he was correct (Krause et al., 1969).
This realization introduced the concept of opportunistic pathogens, or organisms that typically only cause disease under certain predisposing conditions. Do these organisms encode virulence genes in the traditional sense? That question remains the subject of much controversy among modern microbiologists. Sometimes the answer is yes, but often the so-called virulence genes encoded by these organisms are as unassuming as metabolic genes that allow growth on certain substrates. It just so happens that, in the absence of an immune response, these organisms are free to grow and replicate without restraint. The reaction (or lack thereof) of the host to the pathogen is thus an important component of infection.
The Molecular Version of Koch's Postulates
This controversy led to the development of a molecular version of Koch's postulates (Falkow, 1988, 2004). The original postulates were developed in the 1870s by Robert Koch, a country doctor faced with more farm-related anthrax cases than he could handle. Koch purchased a microscope for his clinic and noticed large organisms swimming in the blood of his infected patients. After isolating the bacteria in a makeshift lab, Koch realized he could elicit anthrax by injecting animals with pure cultures of the bacterium, eventually known as Bacillus anthracis. The doctor's technique—extracting the suspected agent of disease, growing it in pure culture, injecting the purified agent into an animal, and subsequently isolating it from the diseased animal—became known as Koch's postulates. Koch later used this process to identify the causative agents of cholera and tuberculosis, and his postulates laid the groundwork for modern studies of virulence.
Today, researchers know that the pathogenicity of many pathogens—pathogens that fail to fulfill the postulates and otherwise might be rejected—plays a causal role in disease and infection. It is important to note that Koch's postulates provide scientists with a framework to guide research. New technologies and information, including the genomic sequences of many of these pathogens, now offer a more complete picture of the requirements of pathogenicity.
Summary
The field of microbial genetics has come a long way since Van Leeuwenhoek's first renderings of microorganisms in the 1600s. Thanks to recent advances in genomics and proteomics, scientists have learned that the genes involved in virulence tend to share similar properties. Many of these genes are encoded on pathogenicity islands, bacteriophages, or insertion sequences nestled within an organism's chromosomal DNA. Other so-called virulence genes can assist with metabolism and thus play an inadvertent role in pathogenicity by allowing otherwise commensal organisms to flourish in warm, moist environments such as the human body. Once identified, these genes can be further examined to reveal the complex pathways between gene expression within virulent microorganisms and the development of disease.
References and Recommended Reading
Elliott, S. J., et al. The complete sequence of the locus of enterocyte effacement (LEE) from enteropathogenic Escherichia coli E2348/69. Molecular Microbiology 28, 1–4 (1998)
Falkow, S. Molecular Koch's postulates applied to microbial pathogenicity. Reviews of Infectious Diseases 10, S274–S276 (1988)
———. Molecular Koch's postulates applied to bacterial pathogenicity—A personal recollection 15 years later. Nature Reviews Microbiology 2, 67–72 (2004) doi:10.1038/nrmicro799 (link to article)
Galán, J. E., & Wolf-Watz, H. Protein delivery into eukaryotic cells by type III secretion machines. Nature 444, 567–573 (2006) doi:10.1038/nature05272 (link to article)
Kaper, J. B., et al. Pathogenic Escherichia coli. Nature Reviews Microbiology 2, 123–140 (2004) doi:10.1038/nrmicro818 (link to article)
Krause, W., Matheis, H., & Wulf, K. Fungaemia and funguria after oral administration of Candida albicans. Lancet 1, 598–599 (1969)
Moon, H. W., et al. Attaching and effacing activities of rabbit and human enteropathogenic Escherichia coli in pig and rabbit intestines. Infection and Immunity 41, 1340–1351 (1983)
Perna, N. T., et al. Genome sequence of enterohaemorrhagic Escherichia coli O157:H7. Nature 409, 529–533 (2001) doi:10.1038/35054089 (link to article)
Robinson, C. M., et al. Shiga toxin of enterohemorrhagic Escherichia coli type O157:H7 promotes intestinal colonization. Proceedings of the National Academy of Sciences 103, 9667–9672 (2006) doi:10.1073/pnas.0602359103
World Health Organization. World Health Report 2004 (Geneva, World Health Organization, 2004)










