


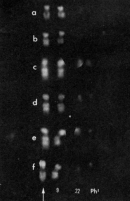
 Figure 1
Figure 1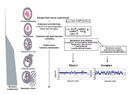















« Prev Next »
In 1960, Peter Nowell and David Hungerford discovered the first chromosomal abnormality associated with cancer using cytogenetics (Nowell & Hungerford, 1960). Specifically, they identified an abnormal minute chromosome that was present in patients with chronic myeloid leukemia, a form of cancer that causes unrestricted growth of myeloid cells in the bone marrow. Nowell and Hungerford named the minute chromosome the Philadelphia chromosome, and the alteration associated with this chromosome was presumed to be a deletion. Later, in 1973, Janet Rowley used new cytogenetic techniques, namely quinacrine fluorescence and G-banding, to examine patients' karyotypes, and in doing so, she discovered that the Philadelphia chromosome actually formed from a specific translocation of DNA between chromosomes 9 and 22. This translocation shortened chromosome 22 to form the Philadelphia chromosome (Ph1), while simultaneously lengthening chromosome 9 (9q+), as shown in Figure 1 (Rowley, 1973). But how does this DNA rearrangement trigger leukemia? It turns out that the translocation leads to the formation of an abnormal, fused gene called bcr-abl, which codes for an aberrant, new protein. This abnormal protein functions as a kinase molecule, which is constitutively, or always, active and can, in turn, activate cell cycle controlling proteins and enzymes (Trask, 2002). This interferes with normal cell cycle regulation and allows cells that express the aberrant protein to divide more rapidly. Such unrestricted cell growth can result in cancer.
Since the discovery of this particular chromosomal rearrangement, thousands of other chromosomal aberrations have been determined to be associated with cancer (Mitelman, 2005). These chromosomal changes are the signature of gene deregulation in cancer and lead to instability of the genome (Albertson et al., 2003). Chromosomal changes are highly variable in different cancers, and the resultant phenotypic effects are equally variable.
Types of Chromosomal Changes
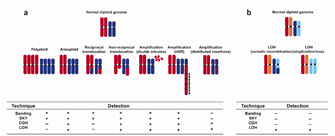
© 2003 Nature Publishing Group Albertson, D. G. et al. Chromosome aberrations in solid tumors. Nature Genetics 34, 370. All rights reserved. 
Scientists have hypothesized that the primary pathogenetic changes in cancer result from balanced rearrangements, while the secondary changes that occur during cancer progression are from unbalanced changes (Mitelman, 2005). Cancer is a multistep, progressive disease, and early chromosomal changes provide the cell with a proliferative advantage. Often, these changes hijack or interfere with the normal cellular control mechanisms by disrupting proto-oncogenes and tumor suppressor genes and allowing additional changes to occur in the genome. Different types of chromosomal changes and the cytogenetic methods commonly used to identify them are shown in Figure 2 (Albertson et al., 2003).
Cancer cells generally gain multiple types of chromosomal aberrations during tumor progression, including rearrangements, deletions, and duplications. As a result, the genome becomes progressively more unstable. Chromosomal Rearrangements
Chromosomal rearrangements can lead to cancer either by forming a hybrid gene or by causing disregulation of a gene. Recall the story of the Philadelphia chromosome, which is formed due to a rearrangement that creates the hybrid bcr-abl gene. The aberrant protein coded for by the hybrid gene accelerates cell division and is associated with chronic myeloid leukemia.
Hybrid genes fuse from portions of two different genes at the rearrangement breakpoints. These rearrangements can be detected by scientists using fluorescence in situ hybridization (FISH). So far, more than 200 of these fusions have been identified (Mitelman, 2005), and they often involve oncogenes, such as MLL, RET, and EWSR1. Sometimes, the same hybrid gene is present in multiple types of cancers, indicating that such genes are not specific to particular cancer types, but rather that these genes might initiate cancer progression in a variety of different tissues (Mitelman, 2005).
Disregulation generally leads to a gene's overexpression or provides a proliferative advantage. For example, the regulatory element of a constitutively active gene (such as an immunoglobulin gene) might be transposed, resulting in activation of a normally silent gene (Mitelman, 2005). Mutations in human DNA repair genes, which maintain genomic integrity, can also deregulate normal cellular processes and allow additional proliferation mutations to occur (Van Gent et al., 2001). Disregulation of normal genes can therefore contribute to the conversion of normal cells into cancerous cells.
Deletions and Duplications
In cancers such as Wilms' tumor and retinoblastoma, gene deletions or inactivations are responsible for initiating cancer progression (Mitelman, 2005). In fact, inactivation of tumor suppressor genes is associated with many types of cancer, as chromosomal regions associated with tumor suppressors are commonly deleted or mutated. For example, deletions, inversions, and translocations are commonly detected in chromosome region 9p21 in gliomas, non-small-cell lung cancers, leukemias, and melanomas (Cairns et al., 1995). These chromosomal changes inactivate a tumor suppressor called cyclin-dependent kinase inhibitor 2A (Cairns et al., 1995). Along with these deletions of specific genes, large portions of chromosomes can also be lost. For instance, chromosomes 1p and 16q are commonly lost in solid tumor cells (Albertson et al., 2003).
Gene duplications and increases in gene copy numbers can also contribute to cancer and can be detected with transcriptional analysis or copy number variation arrays. For example, the chromosomal region 12q13-q14 is strikingly amplified in many sarcomas. This chromosomal region encodes a binding protein called MDM2, which is known to bind to a tumor suppressor called p53. When MDM2 is amplified, it prevents p53 from regulating cell growth, which can result in tumor formation (Oliner et al., 1992).
Additionally, certain breast cancers are associated with overexpression and increases in copy number of the ERBB2 gene, which codes for human epidermal growth factor receptor 2 (Pollack et al., 1999). Indeed, the presence of a high number of ERBB2 copies has been found to be associated with aggressive forms of breast cancer (Peiró, 2004). Therefore, measuring the ERBB2 copy number can provide a diagnostic tool for breast cancer and other cancers. Along with these amplifications of specific genes, gains in chromosomal number, such as chromosomes 1q and 3q, are also associated with increased cancer risk (Albertson et al., 2003).
Mutations in the genes necessary for DNA repair can additionally lead to rearrangements and duplications. For example, if a gene involved with chromosomal segregation is mutated, duplications and deletions are more likely. Furthermore, the accurate sorting and segregation of chromosomes during mitosis requires the activity of many gene products (proteins). Defects in the genes controlling the mitotic surveillance mechanisms necessary for chromosomal sorting can lead to chromosome instability and abnormalities in the number of chromosomes (polyploidy and aneuploidy), which can, in turn, lead to tumorigenesis. Another cause of genome instability and chromosomal aberrations in tumors is the presence of abnormal centromeres, which can lead to abnormal mitotic events with multiple spindles and result in the abnormal loss of chromosomes (Nigg, 2002).
Fragmentary Knowledge: Remaining Challenges
So far, technical methods for detecting cytogenetic changes have favored detection of hematological cancers, or cancers of the blood, such as leukemias; however, the majority of human cancers are solid tumors. Balanced chromosomal changes have been detected in 30% of acute leukemias but only in 5% of epithelial cancers (Mitelman, 2005). This is because hematological cancers can be detected earlier, when the cells have fewer cytogenetic changes. In contrast, solid tumors are often diagnosed later, after a multitude of unbalanced chromosomal mutations have taken place.
Mutations that provide growth or other advantages to cancer cells accumulate at different times during cancer progression, so cells in solid tumors are generally heterogeneous when the cancer is detected. A schematic illustration of epithelial tumor progression is shown in Figure 3 (Albertson et al., 2003). A single mutation in a single cell can allow the cell to escape from its normal tissue organization. An example is the loss of the adenomatous polyposis coli (APC) tumor suppressor gene, which occurs in many colorectal cancers (Fearon & Vogelstein, 1990). Additional mutations in genes such as RAS, CCND, CMYC, CDKN1A, or RB1 can allow a cell to deregulate its normal cell cycle controls, to live longer, and to increase its proliferation (Albertson et al., 2003). Cancer is clonal in origin and begins from mutations like these within a single cell that provide an environment for abnormal cell growth. As a tumor develops, the genome grows more unstable and complex. The telomeres can erode, centromeres might be duplicated, and double-stranded breaks might occur as cancer development progresses. Eventually, the instability of the genome results in more complicated karyotypes as a cancer becomes invasive and metastatic (Albertson et al., 2003).
This complexity can complicate the process of identifying the critical early mutations that promoted the development of the cancer and genomic instability. In fact, to identify common early changes, it is necessary to characterize many clonal lines and identify common mutations. These mutations and aberrations can be identified using modern cytogenetic approaches, including fluorescence in situ hybridization, PCR-based screening, copy number measurements, and comparative genomic hybridization. The common mutations are more likely to be relevant early in cancer development. Therefore, identification of these mutations is critical in order to consistently diagnose cancer at early stages and provide targeted therapies.
References and Recommended Reading
Albertson, D. G., et al. Chromosome aberrations in solid tumors. Nature Genetics 34, 369–376 (2003) doi:10.1038/ng1215 (link to article)
Cairns, P., et al. Frequency of homozygous deletion at p16/CDKN2 in primary human tumours. Nature Genetics 11, 210–212 (1995) (link to article)
Fearon, E. R., & Vogelstein, B. A genetic model for colorectal tumorigenesis. Cell 61, 759–767 (1990)
Mitelman, F. Cancer cytogenetics update 2005. Atlas of Genetics and Cytogenetics in Oncology and Haematology March 2005
Nigg, E. A. Centrosome aberrations: Cause or consequence of cancer progression? Nature Reviews Cancer 2, 815–825 (2002) doi: 10.1038/nrc924 (link to article)Nowell, P., & Hungerford, D. A minute chromosome in human chronic granulocytic leukemia. Science 132, 1497 (1960)
Oliner, J. D., et al. Amplification of a gene encoding a p53-associated protein in human sarcomas. Nature 358, 80–83 (1992) doi:10.1038/358080a0 (link to article)
Peiró, G., et al. Analysis of HER-2/neu amplification in endometrial carcinoma by chromogenic in situ hybridization: Correlation with fluorescence in situ hybridization, HER-2/neu, p53, and Ki-67 protein expression, and outcome. Modern Pathology 17, 277–287 (2004) doi:10.1038/modpathol.3800006
Pollack, J. R., et al. Genome-wide analysis of DNA copy-number changes using cDNA microarrays. Nature Genetics 23, 41–46 (1999) doi:10.1038/12640 (link to article)
Rowley, J. D. A new consistent chromosomal abnormality in chronic myelogenous leukemia identified by quinacrine fluorescence and Giemsa staining. Nature 243, 290–293 (1973) doi:10.1038/243290a0 (link to article)
Trask, B. J. Human genetics and disease: Human cytogenetics—46 chromosomes, 46 years and counting. Nature Reviews Genetics 3, 769–778 (2002) doi:10.1038/nrg905 (link to article)
Van Gent, D. C., et al. Chromosomal stability and the DNA double-stranded break connection. Nature Reviews Genetics 2, 196–206 (2001) doi:10.1038/35056049 (link to article)









