
Abstract
Chronic systemic inflammation is one of the hallmarks of the aging immune system. Here we show that activated T cells from older adults contribute to inflammaging by releasing mitochondrial DNA (mtDNA) into their environment due to an increased expression of the cytokine-inducible SH2-containing protein (CISH). CISH targets ATP6V1A, an essential component of the proton pump V-ATPase, for proteasomal degradation, thereby impairing lysosomal function. Impaired lysosomal activity caused intracellular accumulation of multivesicular bodies and amphisomes and the export of their cargos, including mtDNA. CISH silencing in T cells from older adults restored lysosomal activity and prevented amphisomal release. In antigen-specific responses in vivo, CISH-deficient CD4+ T cells released less mtDNA and induced fewer inflammatory cytokines. Attenuating CISH expression may present a promising strategy to reduce inflammation in an immune response of older individuals.
Similar content being viewed by others


Main
Characteristic hallmarks of immune aging are a decline in adaptive immunity1,2,3, clinically manifesting as increased susceptibility to infections and decreased efficacy of vaccinations, in combination with inflammaging, a low-grade systemic inflammatory state that contributes to widespread tissue dysfunction, frailty and premature death4,5. Inflammatory mediators are produced by a variety of mechanisms, some of which are controlled by T cells, suggesting a causal relationship between changes in the adaptive immune system and inflammaging. The aged T-cell repertoire is shaped by an accumulation of effector T-cell populations that produce proinflammatory cytokines, in part in response to chronic viral infections such as Epstein–Barr virus and cytomegalovirus6. Some T-cell populations, such as terminally differentiated effector memory cells, have acquired innate cell-like properties, enabling them to respond to stimuli other than antigen to produce interferon gamma (IFN-γ) and other cytokines7,8,9.
This proinflammatory T-cell state is not limited to established immunity and effector memory cells. Naive T cells from older adults have a propensity to differentiate into effector T cells due to sustained mTORC1 pathway activation and induction of transcription factor networks dominated by BATF, BLIMP1 and RUNX rather than TCF1 (refs. 10,11). Activation of DNA damage responses in T cells from older adults due to telomere shortening, replication stress and loss of DNA repair molecules further contribute to this inflammatory state12,13,14,15. Most but not all of these T-cell-derived effector molecules are cytokines. A recent study has identified granzyme K, produced by a CD8+ T-cell population in older mice and human adults, as an inducer of inflammatory mediators in senescent cells, also called senescence-associated secretory phenotype16.
In addition to the baseline steady state of inflammatory mediator production in the absence of obvious exogenous stimuli, inflammaging also manifests as excess inflammation in an acute immune response. When occurring in the context of vaccination, it is frequently associated with reduced efficacy in older adults17,18,19,20; conversely, inhibition of inflammation can improve vaccine efficacy21. Excess inflammation occurring in the context of an acute infection in older individuals can result from increased viral load due to reduced or delayed T-cell responses but can also reflect an active mechanism by the responding T-cell population. In general, the increased inflammatory response is not beneficial for the infected host but is associated with excessive tissue injury. As a recent prime example, hyperinflammation can be induced by SARS-CoV-2 infections and is then positively correlated with disease severity and death, including in older adults22,23. The underlying mechanisms have been unresolved although it is remarkable that the disease is associated with severe lymphopenia, reminiscent of the strongly inflammatory lymphocyte pyroptosis in patients with HIV24. T-cell pyroptosis due to mitochondrial DNA (mtDNA)-induced inflammasome activation also contributes to the inflammation in rheumatoid arthritis25. In addition, T-cell responses from older adults have been shown to be associated with increased secretion of granzyme B-containing exosomes, increased DNA damage responses and increased cell death from purinergic signals that all may contribute to inflammation13,26,27.
In this Article, we describe that activated CD4+ T cells from older adults release mtDNA, which is known as a powerful inflammatory mediator. Activation of naive CD4+ T cells from older adults resulted in the increased expression of cytokine-inducible SH2-containing protein (CISH) due to failed repressive action of TCF1. CISH facilitated proteasomal degradation of the V-type proton ATPase catalytic subunit A (ATP6V1A), a component of the vacuolar ATPase that mediates acidification of lysosomes. Reduced ATP6V1A protein prevented lysosomal clearance of autophagosomes and multivesicular bodies (MVBs), leading to intracellular amphisome accumulation and subsequent extracellular release of their cargo. In murine infection and immunization models, CISH silencing in antigen-specific T cells reduced serum mtDNA and inflammatory serum cytokines, while improving antibody production. In conclusion, targeting CISH expression in T-cell responses is a promising strategy to reduce inflammaging and boost immunity in older individuals.
Results
Increased CISH expression in naive T cells from older adults
mtDNA is appreciated as an important inflammatory mediator that is mainly produced by innate immune cells but also other cell types upon activation and cellular stress. T-cell activation is associated with extensive mitochondrial biogenesis and upregulation of oxidative phosphorylation. To screen for inflammatory mediators produced by activated T cells, we determined mtDNA in their supernatants. We found increased concentrations in cultures of activated naive CD4+ T cells from older compared to young adults (Fig. 1a), independent of sex (Extended Data Fig. 1a). Cytoplasmic mtDNA is degraded by DNase II, a lysosomal DNase28. Consistent with this clearance function of lysosomes, chloroquine (CQ) and bafilomycin A1 (Baf) promoted the release of mtDNA into the culture supernatants (Extended Data Fig. 1b). Lysosomal acidification (Extended Data Fig. 1c) and proteolytic activity (Fig. 1b) were reduced in activated CD4+ T cells from older compared to younger adults. To identify differentially expressed genes that could account for the lysosomal dysfunction, we reanalyzed previously published RNA-seq and ATAC-seq data of naive CD4+ T cells from young and old adults in the first 48 hours (h) after activation. Cytoscape gene network analysis identified CISH as one of the top genes, multiple regulatory regions of which were more accessible, and the gene was more transcribed in cells from older individuals (Fig. 1c). CISH has been shown to be an important negative regulator of endolysosomal acidification29. Indeed, silencing of CISH in naive CD4+ T cells from older individuals reduced activation-induced mtDNA release (Fig. 1d and Extended Data Fig. 1d). Kinetic analysis of the human naive CD4+ T-cell transcriptome after stimulation showed an induction of CISH transcripts after activation that was more pronounced with older age (Fig. 1e). The age-associated differences in CISH expression persisted at least until day 5 after activation (Fig. 1f). They were also seen for naive CD8+ T cells but not for activated B cells (Extended Data Fig. 1e). Activated monocytes did not express CISH. Consistent with the transcriptomic data, protein expression of CISH increased after activation and was significantly higher in activated T cells from older adults as shown by immunoblotting and flow cytometry (Fig. 1g).

a, mtDNA copies in supernatants of day-3-stimulated naive CD4+ T cells from eight 20–35 (Y) and fourteen 65–85-year-old (O) adults. b, Lysosomal proteolytic activities determined by flow cytometry of DQ-BSA-treated, day-3-stimulated naive CD4+ T cells comparing eight young and eight older adults. c, Cytoscape network of genes with differential transcript expression and chromatin accessibility comparing day-2-activated naive CD4+ T cells from young and older adults (SRA PRJNA757466). Colors indicate higher (magenta) or lower (cyan) transcripts in the older adults and circle sizes indicate the number of differentially accessible sites associated with the gene. d, mtDNA copies in the supernatants of day-3-stimulated naive CD4+ T cells from five older individuals after control or CISH silencing. e,f, CISH transcripts from SRA PRJNA757466 at the indicated time points11 (e) and naive CD4+ T cells from 12 young and 12 older individuals 5 d after stimulation (f). g, CISH protein levels at indicated time points (left, n = 4) and from day-5-stimulated naive CD4+ T cells (right, n = 6) from young and older individuals. MFI, mean fluorescence intensity. h, Volcano plot of fold differences of TCF1 target gene expression in stem-like versus exhausted mouse CD8+ T cells from PRJNA546023 (ref. 30). TCF1 target genes were obtained from published chromatin immunoprecipitation followed by sequencing66 (GSE52070). Comparisons by two-sided robust empirical Bayes-moderated t-test. FC, fold change. i, TCF1 protein expression in day-5-stimulated naive CD4+ T cells comparing 12 young and 13 older adults. j, mtDNA copies in supernatants of day-3-stimulated naive CD4+ T cells from four young adults after control or TCF7 silencing. k,l, TCF1 protein, CISH transcripts (k) and CISH protein (l) in day-3-stimulated naive CD4+ T cells from four young adults after TCF7 silencing. m,n, TCF1 protein, CISH transcripts (m) and CISH protein (n) in day-3-stimulated naive CD4+ T cells from four older adults after indicated treatment. Data are presented as mean ± s.e.m. Comparison by two-tailed unpaired (a,b,e–g,i) and two-tailed paired t-test (d,j,k–n). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
In exploring upstream regulators of CISH, we found that CISH is a target gene of TCF1 as suggested by transcriptome data (accession no. PRJNA546023) comparing stem-like (TCF1+) versus exhausted (TCF1–) mouse CD8+ T cells (Fig. 1h)30. TCF1 declines upon T-cell activation (Extended Data Fig. 1f) and is significantly lower in activated T cells from older adults (Fig. 1i). Silencing of TCF7 (encoding TCF1) in activated naive CD4+ T cells from young individuals promoted extracellular mtDNA release to the extent seen with older adults, suggesting that reduced TCF1 accounted for the age-associated increase (Fig. 1j). Silencing of TCF7 in activated naive CD4+ T cells from young adults reduced lysosomal acidification and proteolytic activities (Extended Data Fig. 1g), indicating that TCF1 prevents mtDNA release through regulating lysosomal activity. Stimulation of the WNT pathway by a GSK3 inhibitor resulting in increased TCF1 expression restored lysosomal acidification and lysosomal proteolytic activities and prevented mtDNA release by activated T cells from older adults (Extended Data Fig. 1h,i). As GSK3 inhibition has broad effects in addition to upregulating WNT signaling and TCF1, we directly confirmed that CISH transcription is repressed by TCF1. We performed TCF7 silencing in naive CD4+ T cells from young adults and found increased CISH transcript and protein expression (Fig. 1k,l). Conversely, forced overexpression of TCF7 reduced CISH protein in activated T cells from older adults (Extended Data Fig. 1j). Moreover, GSK3 inhibition reduced transcription (Fig. 1m) and protein of CISH (Fig. 1n).
CISH impairs lysosome function by promoting ATP6V1A degradation
To examine whether the increased CISH expression accounts for impaired lysosomal activities, we performed CISH silencing in naive CD4+ T cells from older adults and found increased lysosomal acidification and proteolytic activities (Fig. 2a). Silencing of CISH in T cells from young adults also improved lysosomal acidification but to a lesser extent (Extended Data Fig. 2a). In contrast, CISH overexpression in activated T cells from young adults reduced both, similar to direct silencing of ATP6V1A (Fig. 2b).

a,b, Naive CD4+ T cells were transfected with control, CISH siRNA (a) or ATP6V1A siRNA (b) and activated for 3 d, n = 4. Alternatively, cells were transfected with pCMV6 control vector or CISH vector (b) and activated for 3 d. Lysosomal acidification (top) and proteolytic activities (bottom) were determined by flow cytometry-based analysis of LysoSensor and DQ-BSA-treated cells, respectively. c, Protein expression of ATP6V1A and CISH after CISH silencing, n = 3. d, ATP6V1A protein expression in day-3-stimulated naive CD4+ T cells of older (left) and young (right) individuals after MG132 treatment (1 μM). e, Naive CD4+ T cells were transfected with control or CISH siRNA and activated for 3 d, the last 8 h in the presence of 1 μM MG132. ATP6V1A was immunoprecipitated (IP). ATP6V1A and β-actin protein levels in whole-cell lysates (top). Precipitates were analyzed for ubiquitinated proteins and CISH (bottom). One representative of two experiments. f, Control or CISH-silenced, day-3-stimulated naive CD4+ T cells were treated with CHX (5 μg ml−1) to inhibit de novo ATP6V1A synthesis. Total ATP6V1A protein normalized to β-actin expression is shown relative to non-treatment. g, ATP6V1A protein expression of day-3-stimulated naive CD4+ T cells comparing eight young and eight older individuals. h, mtDNA copies in the supernatants of day-3-stimulated naive CD4+ T cells from five young individuals after ATP6V1A silencing. Data are presented as mean ± s.e.m. Comparison by two-tailed paired t-test (a–c,h) or two-tailed unpaired t-test (g). *P < 0.05, **P < 0.01.
CISH has been reported as a scaffolding protein that promotes ubiquitination of bound proteins and their subsequent proteasomal degradation31,32. Previous studies have found CISH to bind ATP6V1A, which is part of the V-ATPase multi-subunit complex that regulates lysosomal function and in particular luminal acidification29. We explored whether the effects of CISH on lysosomal function was due to regulating ATP6V1A protein levels and found that CISH silencing increased ATP6V1A protein expression (Fig. 2c). To determine whether CISH regulates proteasomal degradation of ATP6V1A, we inhibited proteasome activity with MG132. ATP6V1A protein levels increased in activated T cells from older adults after proteosome inhibition, but much less so in T cells from younger adults (Fig. 2d). Under MG132 treatment, CISH silencing no longer showed any effects on ATP6V1A protein levels (Fig. 2e), suggesting that CISH regulates ATP6V1A protein through the proteasome pathway. Immunoprecipitation of ATP6V1A from MG132-treated cells identified CISH complexed to ATP6V1A (Fig. 2e). Moreover, CISH silencing reduced ATP6V1A ubiquitination, suggesting that the CISH–ATP6V1A interaction facilitated ubiquitination (Fig. 2e). Confocal imaging confirmed colocalization of CISH and ATP6V1A in the cytoplasm (Extended Data Fig. 2b). ATP6V1A half-life in CISH-silenced naive CD4+ T cells was prolonged compared to control-silenced cells (Fig. 2f). Consistent with increased expression of CISH, day-3-activated naive CD4+ T cells from older adults had lower ATP6V1A protein compared to young individuals (Fig. 2g). No age-associated change in proteasome activity was observed, excluding the possibility that housekeeping proteasomal activities contributed to age-related changes of ATP6V1A protein levels (Extended Data Fig. 2c). While silencing of CISH in T cells from older adults reduced mtDNA release (Fig. 1d), silencing of ATP6V1A in T cells from young adults promoted releasee (Fig. 2h and Extended Data Fig. 2d). Taken together, these data document that CISH binds to ATP6V1A facilitating its ubiquitination and subsequent proteasomal degradation and thereby contributes to lysosome dysfunction with age.
CISH impairs autophagy in naive T cells from older adults
The lysosome is a terminal degradation center for autophagy, and lysosomal dysfunction impairs autophagic flux33,34. After activation, T cells from older adults that had increased CISH and lower ATP6VA1 had higher baseline of LC3B-II, an autophagosome marker that is degraded in lysosomes, whereas LC3B-I was not different (Fig. 3a,b). After adding Baf or CQ to inhibit lysosomal degradation, LC3B-II protein increased to the same level in T cells from young and older adults (Fig. 3a,b), consistent with an impaired lysosomal degradation of LC3B-II in T cells from older individuals. To directly determine autophagic flux, we lentivirally transduced naive CD4+ T cells from young and older adults with the RFP–LC3 or RFP–GFP–LC3 tandem fluorescent probes and determined the frequencies of fluorescent cells by cytometry. LC3 targets the probe to the autophagosome, the fluorescence of GFP is quenched when exposed to low pH, whereas RFP is stable. Similar to LC3B-II, frequencies of RFP-fluorescing cells were increased in T cells from older adults, indicating reduced degradation (Fig. 3c). In studies with the RFP–GFP–LC3, the ratio of the acid-labile GFP to the more-resistant RFP fluorescence was increased indicating reduced autophagic flux in activated T cells from older adults (Fig. 3c). Manipulation of CISH expression recapitulated age-associated changes of LC3B-II. As shown in Fig. 3d, overexpression of CISH increased LC3B-II baseline while not affecting LC3B-I or LC3B-II after CQ treatment. Conversely, CISH silencing in T cells from older individuals reduced the LC3B-II baseline consistent with improved autophagic flux (Fig. 3e). cGAS-Sting signaling was not involved in the CISH-induced impairment in autophagic flux. Treatment with the cGAS inhibitor G140 did not prevent the reduction of mtDNA release by CISH silencing (Fig. 3f).

a, Naive CD4+ T cells from young and older individuals were activated for 3 d with the last 6 h in the presence or absence of 50 nM Baf. Cells were probed for LC3B by western blotting; results for LC3B-II and LC3B-I in samples with and without Baf treatment are shown, n = 4. b, Naive CD4+ T cells from three young and three older individuals were activated for 3 d, the last 6 h in the presence or absence of 10 μM CQ. Cells were probed for LC3B by immunoblotting; intensities for LC3B-II and L3B-I are shown. c, Naive CD4+ T cells were lentivirally transduced with an RFP–LC3 or RFP–GFP–LC3 vector. Frequencies of RFP+ in RFP–LC3 and GFP+RFP+ in RFP–GFP–LC3-transduced day-3-stimulated naive CD4+ T cells from six young and six older individuals. Autophagic flux was determined as GFP:RFP ratio. d,e, Naive CD4+ T cells were transfected with pCMV6 control or CISH-expressing vector (d) or control or CISH siRNA (e) and stimulated for 3 d, the last 6 h in the presence or absence of 10 μM CQ. LC3B was immunoblotted, n = 3. f, mtDNA copies in the supernatants of day-3-stimulated naive CD4+ T cells from five older individuals after CISH silencing ± treatment with the cGAS inhibitor G140 at indicated concentrations. Data are presented as mean ± s.e.m. Comparison by two-tailed unpaired t-test (a–c), two-tailed paired t-test (d,e) and one-way ANOVA followed by Tukey’s multiple comparisons test (f). *P < 0.05; **P < 0.01; ***P < 0.001; NS, not significant.
CISH impairs the clearance of autophagosomes and autolysosomes
To explore morphological correlates of autophagic events, we performed transmission electron microscopy (TEM) to visualize and quantify autophagosomes and autolysosomes. Consistent with reduced autophagic flux, day-3-activated naive CD4+ T cells from older adults had significantly more autophagosomes and autolysosomes than those from young individuals (Fig. 4a). Reducing lysosomal activities in T cells from young individuals by either ATP6V1A silencing (Fig. 4a), CISH overexpression (Fig. 4b) or Baf treatment (Fig. 4c) increased the numbers of autophagosomes and autolysosomes. In contrast, enhancing lysosomal activities in T cells from older adults by CISH silencing reduced their numbers (Fig. 4a) without that the number of lysosomes significantly increased (Extended Data Fig. 3). The accumulation of autophagic vacuoles, including autophagosomes, was much more prominent on day 3 after activation than at the resting state of naive CD4+ T cells from older individuals that has been previously reported35. Taken together, these data indicate that excessive induction of CISH transcription in T-cell responses from older adults impairs autophagic flux, leading to intracellular accumulation of autophagosomes and autolysosomes containing non-degraded cargo.

a, Representative TEM images (top) and quantitative assessments of autophagosome and autolysosome numbers (bottom) in day-3-activated naive CD4+ T cells from five young and five older individuals. CISH was silenced in cells from older participants, ATP6VA1 in cells from young individuals. Image acquisition and analysis were performed by an examiner blinded to the nature of the specimen. Results from 15 sections of a representative old and young individual (left) and mean organelle numbers for each of the donors (right) are shown. Ctrl, control. b, Representative TEM images (top) and quantification (bottom) of autophagosome numbers in day-3-stimulated naive CD4+ T cells from three young adults transfected with pCMV6 control or CISH-expressing vector. c, Representative TEM images (top) and quantitative assessments (bottom) of autophagosome number in day-3-stimulated naive CD4+ T cells from five young adults cultured in the presence or absence of 5 nM Baf. Scale bar, 1 μm. Data are presented as mean ± s.e.m. Comparison by two-tailed unpaired t-test (a–c) and two-tailed paired t-test (b). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
CISH induces MVB expansion in naive CD4+ T cells
In addition to autophagosomes, MVBs are organelles that are joined to lysosomes for degradation of their cargo36. Due to reduced lysosomal activities, day-3-activated naive CD4+ T cells from older adults had increased MVBs compared to cells from young individuals (Fig. 5a). Again, MVB numbers could be modulated by regulating lysosomal activities. Enhancing lysosomal activities in T cells from older adults by CISH silencing reduced the numbers of MVBs. Conversely, inhibiting lysosomal activities in T cells from young individuals by ATP6V1A silencing increased their numbers (Fig. 5a). Single-cell ATAC (scATAC-seq)/(scRNA-seq) (scMultiomics) data from a previous study comparing naive CD4+ T cells from young and older adults 18 h after activation showed an enrichment for the endosomal vacuolar gene pathway in cells of older individuals who had high CISH expression (Fig. 5b). As previously reported, this cluster was enlarged in the older population11. Taken together, increased CISH expression in T cells from older individuals results in an expansion of the late-endosomal compartment.

a, Representative TEM images (top) and quantification of MVB numbers (bottom) in day-3-activated naive CD4+ T cells from five young and five older individuals after indicated gene silencing. Data collection and analysis were conducted in a blinded manner. Fifteen sections were analyzed for each donor. Results from a representative older and young donor (bottom left) and mean numbers of five older and five young donors (bottom right) are shown. Scale bar, 1 μm. Comparison by one-way ANOVA followed by Tukey’s multiple comparison test. *P < 0.05, **P < 0.01. Data are presented as mean ± s.e.m. b, Uniform Manifold Approximation and Projection analysis of integrated scATAC-seq and scRNA-seq data from activated naive CD4+ T cells from young and older adults. CISH transcriptional expression and enrichment scores for the endosomal_vacuolar_pathway module are shown by gradient color. Data are reanalyzed from elsewhere11 under accession no. PRJNA757466.
CISH impairs clearance of damaged mitochondria
TEM images showed higher frequencies of damaged mitochondria in activated T cells from older individuals compared to those from young adults (Fig. 6a). Again, these age-related changes depended on lysosomal activities as modulated by CISH. CISH silencing reduced the frequencies of damaged mitochondria in T cells from older adults. Conversely, ATP6V1A silencing increased their frequencies in T cells from young individuals (Fig. 6a). While we observed increased frequencies of damaged mitochondria in naive CD4+ T-cell responses from older individuals, the total numbers of intracellular mitochondria were not affected by age (Fig. 6a). Moreover, levels of COX IV as determined by immunoblotting were not influenced by age suggesting no change in mitochondrial mass (Extended Data Fig. 4a). For a more quantitative assessment of mitochondrial damage, we used flow cytometry. Cell populations with damaged mitochondria can be identified as displaying lower mitochondrial membrane potential, in particular a population referred to as TMRMloMtGhi cells has been described that combine a low potential with increased mass37. We found that the frequencies of activated CD4+ T cells with low membrane potential increased with age, including but not limited to TMRMloMtGhi cells (Fig. 6b and Extended Data Fig. 4b). These cells were small without evidence of apoptosis (Extended Data Fig. 4b). They had reduced TCF1 expression and higher CISH expression compared to the main population (Fig. 6c). LysoSensor staining and lysosomal proteolytic activity were reduced. TEM studies confirmed a higher frequency of damaged mitochondria in the sorted TMRMloMtGhi compared to the TMRMhiMtGhi population, whereas the total numbers of mitochondria per cells was not different (Fig. 6d). Consistent with our studies on age-associated lysosomal dysfunction, CISH expression was an upstream regulator of accumulation of damaged mitochondria. CISH silencing in day-3-activated naive CD4+ T cells from older individuals reduced the frequencies of the TMRMloMtGhi population to about the same frequencies as in young adults (Fig. 6b). Conversely, lysosome inhibition by ATP6V1A silencing or CISH overexpression increased the frequencies of the TMRMloMtGhi population in T cells from young individuals (Fig. 6e,f). Similar effects were observed for the TMRMloMtGlo population that were also enriched in older adults (Extended Data Fig. 4c) Taken together, these data document an intracellular accumulation of damaged mitochondria in T-cell responses from older adults that was caused by reduced lysosomal activities due to increased CISH expression.

a, Representative TEM images (left) and quantitative plots of total mitochondrial numbers (top) and percentage (bottom) of damaged mitochondria (right) in day-3-activated naive CD4+ T cells from five young and five older individuals after indicated gene silencing. Damaged mitochondria were determined as either no visible cristae, surrounded by a phagophore or located inside an autophagic vacuole. Data collection and analysis were conducted in a blinded manner. Results are shown for sections of representative donors and mean of 15 sections from each donor. Scale bar, 1 μm. b, Frequencies of TMRMloMtGhi population in day-3-activated naive CD4+ T cells from six young and seven older individuals after CISH silencing. c,d, Mitochondrial mass and membrane potential of day-3-activated naïve CD4+ T cells from three older individuals were examined using MtG (100 nM) and TMRM (10 nM), respectively. TMRMhiMtGhi and TMRMloMtGhi population were sorted and analyzed for indicated markers (c). Alternatively, cells were analyzed by TEM (d). Data collection and analysis were conducted in a blinded manner. Twenty sections were analyzed for each sample. Scale bar, 1 μm. e, Frequencies of TMRMloMtGhi population in day-3-activated naive CD4+ T cells from four young individuals after ATP6V1A silencing. f, Frequencies of TMRMloMtGhi population in day-3-activated naive CD4+ T cells from four young individuals transfected with pCMV6 control vector or CISH-expressing vector. Data are presented as mean ± s.e.m. Comparison by one-way ANOVA followed by Tukey’s multiple comparison test (a,b), two-tailed unpaired t-test (d) or two-tailed paired t-test (c,e,f). *P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001; NS, not significant.
CISH promotes amphisomal exocytosis of mitochondria
As described in Fig. 5, activated T cells from older adults have an expanded late-endosomal compartment with accumulation of MVBs that are prone to release exosomes26. In addition to MVBs, T cells from older individuals had an abundance of endosomal vacuoles (Fig. 7a). Again, this age-related change is regulated by lysosomal activity. Enhancing lysosomal activities by CISH silencing in activated T cells from older individuals reduced endosomal vacuole numbers, whereas inhibiting lysosomal activities by ATP6V1A silencing in T cells from young adults increased their numbers (Fig. 7a). These endosomal vacuoles seemed to be distinct from regular MVBs. MVBs showed dozens of intraluminal vesicles (ILVs) with a size ranging from ~50 nm to ~100 nm inside the lumen that were released into the extracellular milieu as exosomes (Fig. 5a). The endosomal vacuoles contained autophagic cargo, double-bilayer membrane-structured autophagosome-like vesicles and damaged mitochondria in the lumen (Fig. 7b), suggesting that they represent amphisomes. Confocal imaging showed accumulation of structures with CD63/LC3B and CD63/COX IV colocalizing in activated T cells from older individuals, but not in CISH-silenced cells or in cells from young adults, indicating again the existence of mitochondria-containing amphisomes in activated T cells from older adults (Extended Data Fig. 5a,b). Frequencies of annexin V+ apoptotic cells were not different in the populations, excluding that amphisome-containing cells were apoptotic (Extended Data Fig. 6a). Moreover, the amphisome-rich T cells from older adult did not display the electron morphology of apoptotic cells, such as condensed and fragmented chromatin and budding of apoptotic bodies (Extended Data Fig. 6b). Consistent with CISH expression being downstream of TCF1, silencing of TCF7 in young adults promoted the intracellular accumulation of amphisomes (Extended Data Fig. 7). Taken together, these data documented the accumulation of damaged mitochondria-containing amphisomes in activated naive CD4+ T cells from older individuals due to increased CISH expression causing lysosomal dysfunction.

a, Representative TEM images (top) and quantitative plots of amphisome numbers (bottom) in day-3-activated naive CD4+ T cells from five young and five older individuals after indicated gene silencing. Image acquisition and analysis were conducted blinded to the nature of the specimen. Results are shown for 15 sections from representative donors (left) and means of 15 sections (right) for each donor. Scale bar, 1 μm. b, Representative TEM images of amphisomes in day-3-activated naive CD4+ T cells from an older individual. Scale bar, 200 nm. c, Representative TEM image showing an intracellular amphisome containing damaged mitochondria and ILVs in a day-3-stimulated naive CD4+ T cell from an older individual and extracellular damaged mitochondria and exosomes next to the cell. Scale bar, 1 μm. d, Representative TEM image showing release of cargo by a day-3-stimulated naive CD4+ T cell from an older individual. Scale bar, 1 μm. e, Proposed longitudinal time course of amphisomal exocytosis as reconstructed from cross-sectional TEM images of day-3-activated naive CD4+ T cells from an older individual. Images in c–e are representative images from the experiments described in a. Scale bar, 200 nm. Mt, mitochondria; Exo, exosome; Amphi, amphisome; PM, plasma membrane; AP, autophagosome; Php, phagophore. Data are presented as mean ± s.e.m. Comparison by one-way ANOVA followed by Tukey’s multiple comparison test (a). *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
Studies of TEM images of T cells from older adults suggested the disposal of amphisome cargo into the extracellular space. Figure 7c shows a cell where the luminal content (exosomes and damaged mitochondria) of an amphisome was not yet completely detached from the cell membrane; another amphisome seemed to be close to undergoing exocytosis. Other cells seemed to have completely released a cargo resembling that in amphisomes (Fig. 7d). In cross-sectional TEM analyses, we identified cells in different states that represented the sequence of events in amphisomal exocytosis: (1) mitophagy induction; (2) MVB and autophagosome contact and fusion; (3) amphisome formation and migration toward the plasma membrane; (4) amphisome and plasma membrane fusion; (5) amphisomal exocytosis of luminal mitochondria; and (6) dispersing damaged mitochondria in the extracellular space (Fig. 7e). Taken together, these data document an amphisome-dependent release of damaged mitochondria that was higher in T-cell responses from older individuals due to increased CISH expression and lysosomal dysfunction.
CISH silencing attenuates mtDNA-induced inflammation in vivo
To investigate whether CISH-dependent extracellular release of mtDNA causes inflammation in vivo, we used the lymphocytic choriomeningitis virus (LCMV) infection mouse model. We retrovirally transduced SMARTA CD4+ T cells with short hairpin RNA (shRNA) specific for Cish (shCish) or control shRNA (shCtrl) and transferred the cells into 5–8-week-old B6 mice followed by acute LCMV infection. Reduced CISH protein expression and increased lysosomal activities after CISH silencing were confirmed before adoptive transfer (Fig. 8a–c). At day 6 after LCMV infection, mice that received Cish-silenced SMARTA CD4+ T cells had reduced serum mtDNA concentrations compared to mice with control-silenced cells (Fig. 8d). TEM images showed reduced numbers of amphisomes in Cish-silenced CD4+ SMARTA cells compared to control cells (Fig. 8e), indicating that the reduced serum mtDNA levels were at least in part due to reduced amphisomal exocytosis. Consistent with reduced serum mtDNA levels, mice receiving Cish-silenced SMARTA CD4+ T cells had lower inflammatory cytokine serum concentrations as well as lower cytokine transcripts in splenocytes compared to mice that received control-silenced cells (Fig. 8f,g). Virus clearance from spleen was not different in mice receiving wild-type or Cish-silenced SMARTA CD4+ T cells (Extended Data Fig. 8a). Also, no difference due to CISH silencing was seen for the differentiation of SMARTA T cells as shown for expression of CD69 and CD44 or type 1 helper T (TH1) cell and follicular helper T (TFH) cell markers (Extended Data Fig. 8b–d). CISH silencing with a second shRNA reproduced the data (Extended Data Fig. 9), excluding off-targets effects.

a–g, shCish or shCtrl retrovirally transduced SMARTA CD4+ T cells were adoptively transferred into B6 mice followed by LCMV infection. Results are pooled from three experiments with 4–10 mice in each group. CISH and ATP6V1A protein expression in transduced cells before adoptive transfer (a). Lysosomal acidification (b) and proteolytic activities (c) in transduced cells before adoptive transfer. Serum mtDNA copies at day 6 after infection (d). Number of amphisomes in sorted SMARTA cells at day 6 (D6) after infection (e). Image acquisition and analysis were conducted in a blinded manner. Representative images and results from 20 sections for each sample. Scale bar, 1 μm. Proinflammatory cytokine concentrations in serum (f). Cytokine transcripts in splenocytes at day 6 after infection (g). h–k, shCish or shCtrl retrovirally transduced naive OT-II CD4+ T cells were adoptively transferred into naïve recipient followed by OVA immunization. Serum mtDNA copies (h), proinflammatory serum cytokine concentrations (i), transcripts in splenocytes (j) and serum anti-OVA IgG1 levels (k) at day 8 after immunization. TNF, tumor necrosis factor. Data are representative of two independent experiments with three mice per group. Data are presented as mean ± s.e.m. Comparison by two-tailed paired t-test (b,c), one-way ANOVA followed by Tukey’s multiple comparison test (d,g,j) or two-tailed unpaired t-test (e,f,h,i,k). *P < 0.05, **P < 0.01; ***P < 0.001; NS, not significant.
To determine whether CISH-dependent inflammation is seen in a non-infectious model, we used ovalbumin (OVA) immunization. We retrovirally transduced OT-II CD4+ T cells with Cish or Ctrl shRNA and transferred the cells into 5–8-week-old B6 mice followed by OVA immunization. Similar to the LCMV model, mice having received Cish-silenced OT-II CD4+ T cells had reduced serum mtDNA and inflammatory cytokine serum concentrations on day 8 after immunization as well as lower proinflammatory gene transcripts in splenocytes compared to mice with control-silenced cells (Fig. 8h–j). Moreover, mice having received Cish-silenced OT-II CD4+ T cells had increased frequencies and absolute numbers of TFH cells in the spleen with better antibody responses as shown by increased serum anti-OVA antibodies (Fig. 8k and Extended Data Fig. 8e).
Discussion
Here, we show that T-cell responses from older adults are proinflammatory due to excessive release of mtDNA and other amphisomal products, thereby assigning T cells a regulatory role in inflammaging that goes beyond cytokine production. At a center stage in this age-associated response pattern is a lysosomal defect due to the accelerated proteasomal degradation of ATP6V1A and the associated failure in lysosomal acidification. Tagging of ATP6V1A for degradation is facilitated by the scaffolding activity of CISH, the transcription of which is induced by T-cell receptor (TCR) activation in older T cells that have reduced activity of the transcriptional repressor TCF1. We have previously reported that replenishment of lysosomes is impaired in activated T cells undergoing proliferation due to a reduced expression of the transcription factor TFEB that controls the transcription of lysosomal genes26. The defect in lysosomal acidification is an independent additional mechanism in T-cell responses from older adults identifying several age-associated changes converging on lysosomal dysfunction. Taken together, T cells from older adults litter their immediate environment with mitochondrial and other cellular products that have been targeted for lysosomal degradation, a defect that can be prevented by silencing CISH.
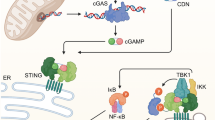
Lysosomal degradation of damaged mitochondria is initiated by the formation of double-membraned autophagosomes that seal the whole mitochondrion or its damaged area followed by fusion with lysosomes to form autolysosomes and ultimately the degradation of mitochondria in the autolysosomal lumen38,39. In some types of cells, before fusion with lysosomes, autophagosomes fuse with late endosomes to form the intermediate/hybrid organelle amphisomes, which contain both late-endosomal and autophagic cargos40. The formation of amphisomes allows autophagosomes to acquire the transport motility from the late endosomes/MVBs, which facilitates their fusion with lysosomes40. In several age-related neurodegenerative diseases, a defect of lysosomal clearance results in the abnormal intracellular accumulation of amphisomes, leading to amphisome/plasma membrane fusion and subsequent amphisome-mediated extracellular secretion of luminal contents40,41. Likewise, we find an intracellular accumulation of amphisomes in activated naive CD4+ T cells from older adults. Due to lysosome dysfunction in these T cells, amphisomes are no longer targeted for degradation but fuse with the plasma membrane and release their luminal contents, including damaged mitochondria to the extracellular milieu. Released damaged mitochondria provide a source of circulating mtDNA, which is one of the most important inducers of inflammaging42,43. mtDNA activates innate recognition receptors, including cGAS-STING and Toll-like receptors, resulting in inflammasome activation and production of type I IFN and proinflammatory cytokines44,45. Previous studies on inflammaging have mostly focused on the cytosolic leakage of mtDNA in innate immune cells upon activation and cellular stress45. Here, we find that secretion of mtDNA is inherent in a normal CD4+ T cell response and heightened with T cells of older adults.
The autophagy–lysosome pathway is essential to maintain organelle homeostasis and thus to prevent age-associated inflammation46,47. Previous studies have shown an age-associated decline of macroautophagy in resting human T cells48,49. The underlying mechanisms for this defect are poorly understood. Transcriptomic analyses so far have failed to identify autophagy-related gene signatures associated with age in T cells50. One possible mechanism is an increase in MAPK p38 activity in selected older T cells that inhibits ATG9 activity51. Previous studies were mostly conducted on basal autophagy in resting cells, which is different from activation-induced autophagy52. Our studies were performed on activated T cells that, in contrast to resting T cells, are anabolic undergoing organelle and mitochondrial biogenesis. Here, the upstream mechanism of increased MVB and amphisome accumulation was a defective clearance function of lysosomes.
Deficiency in TCF1 expression is one hallmark of T-cell aging. One of the target genes of TCF1 is CISH, a member of the SOCS family (Fig. 1h). TCF1 has been shown to bind to the CISH promoter repressing its transcription53. CISH acts as a scaffolding protein by binding ATP6V1A and facilitating its ubiquitination and proteasomal degradation. ATP6V1A is an essential component of the proton pump V-ATPase that maintains lysosomal acidification. Excessive induction of CISH transcription in activated naive CD4+ T cells from older individuals increased ATP6V1A degradation and impaired lysosomal clearance of autophagosomes, thereby leading to intracellular accumulation of damaged mitochondria and amphisomes, resulting in exocytosis of damaged mitochondria. In addition to defective clearance, age-related mitochondrial defects may contribute to their accumulation and aggravate the inflammatory response3,25,54.
In addition to the in vitro data with human cells, we have confirmed a role of CISH in regulating inflammation in murine models of antigen-driven T-cell responses. Increasing the number of antigen-specific T cells by adoptively transferring TCR-transgenic T cells induced an increase in the serum concentrations of mtDNA in the LCMV infection as well as the ovalbumin immunization model. Mice adoptively transferred with antigen-specific Cish-silenced CD4+ T cells had reduced serum mtDNA and proinflammatory cytokine concentrations, confirming that mtDNA was derived from the responding T-cell population and was CISH dependent.
Further studies are needed to assess whether, in addition to aging, increased CISH expression accounts for increased inflammation via mtDNA release in several settings of chronic T-cell activation. CISH has been reported to be highly induced in the chronic LCMV infection model53. CISH is also highly expressed in tumor-infiltrating lymphocytes55. In both models, CISH has been implicated to account for attenuated TCR signaling in exhausted T cells, but it may also contribute to the chronic inflammatory response in these conditions. Of particular interest, increased serum mtDNA derived from T cells may contribute to the inflammatory cytokine release syndrome commonly observed in patients receiving adoptive CAR-T-cell therapy56,57,58. CISH knockout in adoptively transferred T cells is currently explored in a human clinical trial for the treatment of patients with gastrointestinal cancer (https://clinicaltrials.gov/ct2/show/NCT04426669)59. Although the trial was originally conceived to combat exhaustion, our data suggest that it may also show a reduced incidence and severity of cytokine release syndrome.
mtDNA release at the height of the T-cell response will hyperstimulate innate immunity, which may be beneficial to control a viral infection, but also cause tissue damage. In addition, this proinflammatory environment will also impact the adaptive immune response and T-cell differentiation. A previous study found that impaired lysosomal function directs T-cell differentiation toward an inflammatory lineage, exacerbating an inflammatory response60. In contrast to this positive feedback loop, excessive inflammation may also curtail the generation of adaptive immune response17,20,61. Upregulation of PD-L1 on inflammatory macrophages inhibits T cells from older adults that express more PD-1 after activation26,62. Excess type I IFN reduces the number of antigen-specific IFN-γ+ CD4+ T cells. Blockade of type I IFN signaling resulted in increased numbers of IFN-γ+ CD4+ T cells and thus enhanced virus clearance63,64. In support of such an immunosuppressive effect, we found that mice receiving Cish-silenced SMARTA CD4+ T cells had a reduced inflammatory response after acute LCMV infection compared to mice receiving control-silenced cells. A similar improved adaptive immune response was seen in an immunization model, where TFH cell frequencies and serum anti-OVA antibodies were increased after transfer of Cish-silenced OT-II CD4+ T cells.
In summary, we describe that lysosomal function in responding T cells is essential to maintain a healthy immune microenvironment with low inflammation and superior outcome in antibody responses. Impaired lysosomal activities in T-cell responses from older individuals promotes amphisomal exocytosis of intracellular waste, including damaged mitochondria and mtDNA that induces local activation of inflammatory cells. It should be noted that the inflammation described here occurs in an acute immune response of largely healthy, older individuals, such as in an acute infection or a vaccination. Whether the same mechanisms contribute to the chronic subtle systemic inflammation seen in older adults is possible but remains to be seen. Inflammaging is more prevalent in frail individuals and one of the limitations of our study is that we did not study the relationship with frailty. Irrespective of how systemic the process is that we are describing, with the excessive induction of CISH transcription we have identified a key mechanism that can be targeted to reduce T-cell-dependent inflammation and improve adaptive immunity in older individuals.
Methods
Study population and cell isolation
PBMCs were obtained from leukocyte reduction chambers of 57 young (20–35 years old) and 64 older (65–85 years old) healthy blood donors of both sexes, purchased from the Stanford Blood Center and the Mayo Clinic Blood Center. The study was conducted in accordance with the Declaration of Helsinki and approved by the Stanford Institutional Review Board and the Mayo Clinic Institutional Review Board. Samples had been de-identified except for age range and sex. Human naive CD4+ T cells were purified using a CD4+ T-cell enrichment cocktail kit (15062, STEMCELL Technologies), followed by negative selection with anti-CD45RO magnetic beads (19555, STEMCELL Technologies). Human naive CD8+ T cells and naive B cells were purified by using human naive CD8+ T Cell Isolation kit (19258, STEMCELL Technologies) and Human Naive B Cell Isolation kit (17254, STEMCELL Technologies), respectively. Human CD14+ monocytes were isolated by positive selection with CD14 MicroBeads (130050201, Miltenyi Biotec).
Cell culture
Freshly isolated human naive CD4+ T cells or naive CD8+ T cells were activated at a density of 5 × 105 ml−1 with anti-CD3/anti-CD28 Dynabeads (11132D, Thermo Fisher Scientific) in RPMI 1640 (Sigma-Aldrich) supplemented with 10% fetal bovine serum (FBS) and penicillin and streptomycin (100 U ml−1, Thermo Fisher Scientific). Freshly isolated human naive B cells were expanded by using CellXVivo Human B Cell Expansion kit (CDK005, R&D system) for 5 d. Freshly isolated human CD14+ monocytes were stimulated with 20 ng ml−1 M-CSF for 4 days (d) followed by 100 ng ml−1 LPS for 1 d. Freshly isolated mouse naive CD4+ T cells (19765, STEMCELL Technologies) were activated at a density of 2 × 106 ml−1 in plates coated with anti-CD3 (8 μg ml−1; 16-0032-82, eBioscience) and anti-CD28 antibody (8 μg ml−1; 16-0281-82, eBioscience) in culture medium supplemented with interleukin (IL)-2 (10 ng ml−1; 21212, PeproTech).
Transfection
Freshly isolated human naive CD4+ T cells or naive CD8+ T cells were transfected with either SMARTpool negative control siRNA, SMARTpool TCF7 siRNA, SMARTpool CISH siRNA, SMARTpool ATP6V1A siRNA (all from Dharmacon) or pCMV6-Entry Mammalian or Human CISH expression vector (NM_145071, both from Origene) using the Amaxa Nucleofector system and the P3 primary cell Nucleofector kit (Lonza). After transfection, cells were rested for 2 h before being activated by anti-CD3/anti-CD28 Dynabeads for 3 or 5 d. For lentiviral transduction, naive CD4+ T cells were transduced with a lentiviral vector expressing either scrambled control, TCF7, RFP–LC3 or RFP–GFP–LC3 (as the autophagic flux probe). After overnight culture, cells were activated by anti-CD3/anti-CD28 Dynabeads for 3 d.
Immunoblotting
Cells were washed with cold phosphate-buffered saline (PBS) and lysed in commercial RIPA buffer supplemented with phenylmethylsulfonyl fluoride and protease and phosphatase inhibitors (sc-24948, Santa Cruz Biotechnology) for 30 min on ice. Proteins were separated on denaturing 4–15% (15-well) SDS–polyacrylamide gel electrophoresis (4561086, Bio-Rad), transferred onto nitrocellulose or PVDF membrane (1704270, Bio-Rad), and probed with antibodies to TCF1 (C46C7, no. 2206, 1:1,000 dilution), β-actin (D6A8, no. 8457, 1:1,000 dilution), CISH (D4D9, no. 8731, 1:1,000 dilution), LC3B (D11, no. 3868, 1:1,000 dilution), COX IV (3E11, no. 4850, 1:1,000 dilution; all from Cell Signaling Technology) and ATP6V1A (ab199326, Abcam, 1:1,000 dilution). Blotting membranes were developed by incubation with HRP-conjugated secondary antibodies (Cell Signaling Technology, no. 7074, 1:2,500 dilution) followed by treating with the Chemiluminescent Western Blot Detection Substrate (Thermo Fisher Scientific).
Flow cytometry
For cell surface staining, cells were incubated with fluorescently conjugated antibodies in PBS supplemented with 2% FBS at 4 °C or on ice for 30 min. For intracellular protein staining, cells were treated with FOXP3 Fix/Perm Buffer Set (421403, BioLegend) followed by incubation with fluorescently conjugated antibodies at room temperature for 60 min. Dead cells were excluded from the analysis using LIVE/DEAD Fixable Violet Dead Cell Stain kit (Thermo Fisher). Staining for flow cytometry was performed with antibodies to AF-700 CD4 (anti-human: RPA-T4, no. 557922; anti-mouse: RM4-5, no. 557956, 1:100 dilution), FITC CD8 (anti-human: RPA-T8, no. 561948; anti-mouse: 53-6.7, no. 553031; 1:100 dilution) and PE-CXCR5 (anti-mouse: 2G8, no. 561988, 1:50 dilution) all BD Biosciences), TCF1 (anti-human: 7F11A10, no. 655208, 1:100 dilution), AF647 PD-1 (anti-mouse: 29F.1A12, no. 135230, 1:100 dilution), BV421 CD44 (anti-mouse: IM7, no. 103040, 1:100 dilution), PE-Cy7 CD62L (anti-mouse: MEL-14, no. 104418, 1:100 dilution), APC-Cy7 CD69 (anti-mouse: H1.2F3, no. 104525, 1:100 dilution) and AF647 SLAM (anti-mouse: TC15-12F12.2; 1:50 dilution, no. 115918; all from BioLegend). Mouse CXCR5 was stained with biotin-conjugated anti-CXCR5 (2G8, 1:50 dilution, BD Biosciences) followed by staining with allophycocyanin–streptavidin (BD Biosciences). Intracellular CISH was stained with primary anti-CISH (D4D9, no. 8731, 1:100 dilution, Cell Signaling Technology) followed by staining with BV421 anti-rabbit IgG (no. 406410, 1:200 dilution, BioLegend). Cells were analyzed on an LSR Fortessa (BD Biosciences). Flow cytometry data were analyzed using FlowJo.
RNA isolation and quantitative RT–PCR
Total RNA was isolated by using RNeasy Plus Mini kit (74134, QIAGEN) and was reverse transcribed using SuperScript VILO cDNA Synthesis kit (11754, Invitrogen). Quantitative PCR was performed on the ABI 7900HT system (Applied Biosystems) using Power SYBR Green PCR Master Mix (4368706, Thermo Fisher Scientific) according to the manufacturer’s instructions. Oligonucleotide primer sets used in this study are shown in Supplementary Table 1.
Proteasome activity assay
Naive CD4+ T cells were activated for 3 d and homogenized with Pierce IP Lysis Buffer (no. 87787, Thermo Fisher). Proteasomal degradation was quantified by using Proteasome Activity Assay kit (ab107921, Abcam) following the manufacturer’s manual. Briefly, samples were incubated with or without proteasome inhibitor, proteasome substrate and assay buffer for 30 min at 37 °C. Fluorescence was determined on a fluorometric plate reader BioTek Synergy H1 at 350/440 nm.
Lysosomal acidification and proteolytic activity assay
For analyzing lysosomal acidification, day-3-stimulated naive CD4+ T cells were treated with 1 µM LysoSensor (L7535, Thermo Fisher Scientific) diluted in prewarmed culture medium and incubated for 30 min at 37 °C, followed by washes with ice-cold PBS. LysoSensor fluorescence was quantified by flow cytometry. For examining lysosomal proteolytic activities, cells were treated with DQ-BSA (5 µg ml−1; D12050, Thermo Fisher Scientific) diluted in prewarmed culture medium and further incubated at 37 °C for 6 h. After incubation, cells were briefly washed once with ice-cold PBS containing 2% FBS. Fluorescence of cleaved DQ-BSA was analyzed by flow cytometry.
Immunoprecipitation of ATP6V1A
Naive CD4+ T cells were transfected with control or CISH siRNA and activated for 3 d, with the last 8 h in the presence of 1 μM MG132 (Cayman Chemical). Cells were lysed using the Lysed Pierce IP Lysis Buffer (Thermo Fisher) and incubated with anti-ATP6V1A antibody (ab199326, Abcam 1:1,000 dilution) at 4 °C overnight. ATP6V1A was immunoprecipitated by treating the whole-cell lysates with Protein G Agarose (sc-2002, Santa Cruz Biotechnology) at 4 °C for 2 h. After washing five times, immune complexes were boiled for 15 min in Laemmli Sample Buffer (no. 1610747, Bio-Rad). Samples were loaded for electrophoresis and subsequently transferred onto PVDF membranes before immunoblotting for specific markers.
Transmission electron microscopy
In vitro day-3-activated naive CD4+ T cells or sorted mouse SMARTA CD4+ T cells 6 d after LCMV infection were fixed overnight in 1% glutaraldehyde and 4% formaldehyde in 0.1 M phosphate buffer, pH 7.2. Cells were then rinsed for 30 min and postfixed in phosphate-buffered 1% OsO4 for 1 h. After three washes in water, cells were en bloc-stained with 2% uranyl acetate for 30 min. After en bloc staining, cells were rinsed, dehydrated and embedded in Spurr resin. Cells were then polymerized in a 60 °C oven overnight. Sections (60–80 nm) were treated with uranyl acetate and lead citrate and viewed on a JEOL 1400 Plus electron microscope. Images were collected of every cell profile within four grid squares of a single thin section of each sample. Then, 15 or 20 cells were imaged for each donor. Damaged mitochondria were defined as either no visible cristae, surrounded by a phagophore or being located inside an amphisome. Image acquisition and analysis were conducted by an examiner blinded to the type of sample (for example, age and silencing condition).
Human and mouse mtDNA quantification
Absolute human and mouse mtDNA copy numbers were quantified by using the Absolute Human Mitochondrial DNA Copy Number Quantification qPCR Assay kit (no. 8948) and the Absolute Mouse Mitochondrial DNA Copy Number Quantification qPCR Assay kit (no. M8948, both ScienCell), respectively. The human or mouse mtDNA primer sets provided in the kit recognize and amplify a conserved region of human or mouse mtDNA. The reference DNA sample with known human or mouse mtDNA copy number served as a reference for calculating the absolute mtDNA copy numbers of target samples.
Mice, adoptive transfers, LCMV infection and OVA immunization
For the SMARTA/LCMV infection model, naive CD4+ T cells specific to the GP66-77 epitope of LCMV (Armstrong) obtained from 5–8-week-old SMARTA TCR-transgenic mice (CD45.1, a gift from R. Ahmed at Emory University) were activated in plates coated with anti-CD3/anti-CD28 antibody. To avoid a transient silencing effect in vivo by transfection, retroviral vectors expressing either scrambled control or Cish shRNA (5′-CAGTTATACAGTATTTATTTA-3′) were generated using the Plat-E Retroviral Packaging Cell Line (Cell Biolabs) and T cells were transduced on days 1 and 2 after activation. On day 6 after activation, retrovirus-transduced Amcyan-positive SMARTA cells were sorted. Then, 1 × 105 transduced cells were intravenously transferred to 5–8-week-old female C57BL/6 (CD45.2) mice (The Jackson Laboratory). One day after transfer, mice were infected intraperitoneally with 2 × 105 plaque-forming units of LCMV Armstrong. On day 6 after infection, spleen and blood were collected and analyzed. For the OT-II/OVA immunization model, naive CD4+ T cells specific to the chicken ovalbumin 323–339 peptide obtained from 5–8-week-old OT-II TCR-transgenic mice (CD45.1, The Jackson Laboratory) were activated and retrovirally transduced as described for SMARTA cells. On day 6 after activation, retrovirus-transduced Amcyan-positive OT-II cells were sorted; 1 × 105 transduced cells were intravenously transferred to 5–8-week-old female C57BL/6 (CD45.2) mice (The Jackson laboratory). One day after transfer, mice were immunized with 100 μg OVA (Biosearch Technologies) precipitated in 100 μl 5% alum (aluminum potassium sulfate, MilliporeSigma). On day 8 after immunization, spleen and blood were collected and analyzed. Mice were house at a temperature of 23 ± 2 °C, relative humidity of 30–40% and a 12-h light–dark cycle. All animal experiments were approved by, and performed following, the guidelines of the Mayo Clinic Administrative Panel on Laboratory Animal Care Committee.
ELISA for serum cytokines and antibodies
Proinflammatory cytokines (TNF-α, IL-1β and IL-6) serum concentrations were measured using ELISA kits (Thermo Fisher). Briefly, ELISA plates coated with cytokine capture antibody were washed, blocked, and incubated with 50 μl serum samples for 2 h. In parallel, a standard curve was made by incubating twofold serial dilutions of the standards. Plates were then washed and incubated with biotin-conjugated detection antibodies for 1 h. After washing, plates were incubated with streptavidin–HRP. Bound antibodies were detected by adding 3,3′,5,5′-tetramethylbenzidine substrate (TMB Solution) followed by plate reading for absorbance at 450 nm. Cytokine levels were calculated based on a standard curve. OVA-specific IgG1 was measured by using the anti-Ovalbumin IgG1 (mouse) ELISA kit (no. 500830, Cayman Chemical) according to the manufacturer’s instructions.
LCMV viral RNA quantification
Total RNA of the spleen or kidney was isolated and reverse transcribed to complementary DNA using the SuperScript VILO cDNA Synthesis kit (no. 11754050, Thermo Fisher). The kit contains random primers that can reverse-transcribe LCMV viral RNA to cDNA. qPCR was performed by using the previously reported LCMV GP specific primers (forward, 5′-CATTCACCTGGACTTTGTCAGACTC-3′; reverse, 5′-GCAACTGCTGTGTTCCCGAAAC-3′)65. RNA isolated from the spleen from non-infected mice was used as a negative control.
Confocal microscopy
Day-3-stimulated naive CD4+ T cells were fixed in 4% paraformaldehyde, permeabilized with 0.2% Triton X-100 and incubated with primary antibodies to CD63 (no. 556019, BD Pharmingen, 1:100 dilution) together with antibodies to CISH (ab88383, Abcam 1:100 dilution), ATP6V1A (ab199326, Abcam, 1:100 dilution), LC3B (D11, no. 3868, Cell Signaling Technology, 1:100 dilution) or COX IV (3E11, no. 4850, Cell Signaling Technology, 1:100 dilution) at 4 °C overnight. Incubation with secondary antibodies was performed at room temperature for 2 h using Alexa Fluor 488-conjugated AffiniPure donkey anti-rabbit IgG, Cy3-conjugated AffiniPure donkey anti-mouse IgG (Jackson Immuno Research Laboratories), Alexa Fluor 647-conjugated goat anti-rabbit IgG H&L or Alexa Fluor 488-conjugated goat anti-mouse IgG H&L (Abcam), all at 1:500 dilution. The images were analyzed using an LSM 980 microscope system with the ZEN 2010 software (Carl Zeiss) and a ×63 oil immersion objective (Carl Zeiss).
Statistics and reproducibility
Statistical analysis was performed using Prism v.8.0 software. Two-tailed paired or unpaired Student’s t-tests were used for comparing two groups. One-way analysis of variance (ANOVA) with Tukey’s post hoc test was used for multigroup comparisons. Sample sizes to identify differences in mtDNA release, CISH or TCF1 expression between young and older adults were chosen to ensure 80% power with a level of significance of 5% for a difference of their means of 1.5 × s.d. (n ≥ 10). Data distribution was not formally tested but was assumed to be normal. To assess the effect of in vitro intervention (for example, gene silencing and pharmacological inhibition), we used paired testing with sample sizes of 3–5. Sample sizes for the mouse studies were similar to those reported in previous publications. For imaging experiments, we used a sample size of 15–20 sections for each sample with a total of five samples in each group. The investigators examining the images were blinded to the nature of the sample. Experiments were not randomized. P < 0.05 was considered statistically significant. Statistical details and significance levels can be found in figure legends.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Source data are included with this article. Additional primary experimental data of this study are available from the corresponding author upon request. Sequencing data were obtained from the Sequence Read Archive under accession nos. PRJNA546023 and PRJNA757466.
References
Goronzy, J. J. & Weyand, C. M. Mechanisms underlying T cell ageing. Nat. Rev. Immunol. 19, 573–583 (2019).
Nikolich-Zugich, J. The twilight of immunity: emerging concepts in aging of the immune system. Nat. Immunol. 19, 10–19 (2018).
Mittelbrunn, M. & Kroemer, G. Hallmarks of T cell aging. Nat. Immunol. 22, 687–698 (2021).
Franceschi, C., Garagnani, P., Parini, P., Giuliani, C. & Santoro, A. Inflammaging: a new immune-metabolic viewpoint for age-related diseases. Nat. Rev. Endocrinol. 14, 576–590 (2018).
Ferrucci, L. & Fabbri, E. Inflammageing: chronic inflammation in ageing, cardiovascular disease, and frailty. Nat. Rev. Cardiol. 15, 505–522 (2018).
Goronzy, J. J. & Weyand, C. M. Successful and maladaptive T cell aging. Immunity 46, 364–378 (2017).
Pereira, B. I. et al. Sestrins induce natural killer function in senescent-like CD8+ T cells. Nat. Immunol. 21, 684–694 (2020).
Warrington, K. J., Takemura, S., Goronzy, J. J. & Weyand, C. M. CD4+CD28− T cells in rheumatoid arthritis patients combine features of the innate and adaptive immune systems. Arthritis Rheum. 44, 13–20 (2001).
Henson, S. M., Riddell, N. E. & Akbar, A. N. Properties of end-stage human T cells defined by CD45RA re-expression. Curr. Opin. Immunol. 24, 476–481 (2012).
Kim, C. et al. Activation of miR-21-regulated pathways in immune aging selects against signatures characteristic of memory T cells. Cell Rep. 25, 2148–2162 e2145 (2018).
Zhang, H. et al. Aging-associated HELIOS deficiency in naive CD4+ T cells alters chromatin remodeling and promotes effector cell responses. Nat. Immunol. 24, 96–109 (2023).
Kim, C. et al. Histone deficiency and accelerated replication stress in T cell aging. J. Clin. Invest. 131, e143632 (2021).
Li, Y. et al. Deficient activity of the nuclease MRE11A induces T cell aging and promotes arthritogenic effector functions in patients with rheumatoid arthritis. Immunity 45, 903–916 (2016).
Qi, Q. et al. Defective T memory cell differentiation after Varicella zoster vaccination in older individuals. PLoS Pathog. 12, e1005892 (2016).
Lanna, A., Henson, S. M., Escors, D. & Akbar, A. N. The kinase p38 activated by the metabolic regulator AMPK and scaffold TAB1 drives the senescence of human T cells. Nat. Immunol. 15, 965–972 (2014).
Mogilenko, D. A. et al. Comprehensive profiling of an aging immune system reveals clonal GZMK+ CD8+ T cells as conserved hallmark of inflammaging. Immunity 54, 99–115 (2021).
Parmigiani, A. et al. Impaired antibody response to influenza vaccine in HIV-infected and uninfected aging women is associated with immune activation and inflammation. PLoS ONE 8, e79816 (2013).
Muyanja, E. et al. Immune activation alters cellular and humoral responses to yellow fever 17D vaccine. J. Clin. Invest. 124, 3147–3158 (2014).
Berry, M. P. et al. An interferon-inducible neutrophil-driven blood transcriptional signature in human tuberculosis. Nature 466, 973–977 (2010).
Fourati, S. et al. Pre-vaccination inflammation and B-cell signalling predict age-related hyporesponse to hepatitis B vaccination. Nat. Commun. 7, 10369 (2016).
Vukmanovic-Stejic, M. et al. Enhancement of cutaneous immunity during aging by blocking p38 mitogen-activated protein (MAP) kinase-induced inflammation. J. Allergy Clin. Immunol. 142, 844–856 (2018).
Merad, M. & Martin, J. C. Pathological inflammation in patients with COVID-19: a key role for monocytes and macrophages. Nat. Rev. Immunol. 20, 355–362 (2020).
Akbar, A. N. & Gilroy, D. W. Aging immunity may exacerbate COVID-19. Science 369, 256–257 (2020).
Doitsh, G. et al. Cell death by pyroptosis drives CD4 T-cell depletion in HIV-1 infection. Nature 505, 509–514 (2014).
Li, Y. et al. The DNA repair nuclease MRE11A Functions as a mitochondrial protector and prevents T cell pyroptosis and tissue inflammation. Cell Metab. 30, 477–492 (2019).
Jin, J. et al. FOXO1 deficiency impairs proteostasis in aged T cells. Sci. Adv. 6, eaba1808 (2020).
Fang, F. et al. Expression of CD39 on activated T cells impairs their survival in older individuals. Cell Rep. 14, 1218–1231 (2016).
Matsui, H. et al. Cytosolic dsDNA of mitochondrial origin induces cytotoxicity and neurodegeneration in cellular and zebrafish models of Parkinson’s disease. Nat. Commun. 12, 3101 (2021).
Queval, C. J. et al. Mycobacterium tuberculosis controls phagosomal acidification by targeting CISH-mediated signaling. Cell Rep. 20, 3188–3198 (2017).
Jadhav, R. R. et al. Epigenetic signature of PD-1+TCF1+ CD8 T cells that act as resource cells during chronic viral infection and respond to PD-1 blockade. Proc. Natl Acad. Sci. USA 116, 14113–14118 (2019).
Piessevaux, J., De Ceuninck, L., Catteeuw, D., Peelman, F. & Tavernier, J. Elongin B/C recruitment regulates substrate binding by CIS. J. Biol. Chem. 283, 21334–21346 (2008).
Yoshimura, A., Naka, T. & Kubo, M. SOCS proteins, cytokine signalling and immune regulation. Nat. Rev. Immunol. 7, 454–465 (2007).
Yim, W. W. & Mizushima, N. Lysosome biology in autophagy. Cell Discov. 6, 6 (2020).
He, M. X., McLeod, I. X., Jia, W. & He, Y. W. Macroautophagy in T lymphocyte development and function. Front. Immunol. 3, 22 (2012).
Bektas, A. et al. Age-associated changes in human CD4+ T cells point to mitochondrial dysfunction consequent to impaired autophagy. Aging 11, 9234–9263 (2019).
Huotari, J. & Helenius, A. Endosome maturation. EMBO J. 30, 3481–3500 (2011).
Yu, Y. R. et al. Disturbed mitochondrial dynamics in CD8+ TILs reinforce T cell exhaustion. Nat. Immunol. 21, 1540–1551 (2020).
Palikaras, K., Lionaki, E. & Tavernarakis, N. Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat. Cell Biol. 20, 1013–1022 (2018).
Ma, K. et al. Mitophagy, mitochondrial homeostasis, and cell fate. Front. Cell Dev. Biol. 8, 467 (2020).
Ganesan, D. & Cai, Q. Understanding amphisomes. Biochem. J. 478, 1959–1976 (2021).
Eitan, E., Suire, C., Zhang, S. & Mattson, M. P. Impact of lysosome status on extracellular vesicle content and release. Ageing Res. Rev. 32, 65–74 (2016).
Franceschi, C., Garagnani, P., Vitale, G., Capri, M. & Salvioli, S. Inflammaging and ‘Garb-aging’. Trends Endocrinol. Metab. 28, 199–212 (2017).
Pinti, M. et al. Circulating mitochondrial DNA increases with age and is a familiar trait: implications for “inflamm-aging”. Eur. J. Immunol. 44, 1552–1562 (2014).
Riley, J. S. & Tait, S. W. Mitochondrial DNA in inflammation and immunity. EMBO Rep. 21, e49799 (2020).
Miller, K. N. et al. Cytoplasmic DNA: sources, sensing, and role in aging and disease. Cell 184, 5506–5526 (2021).
Zhang, H., Puleston, D. J. & Simon, A. K. Autophagy and immune senescence. Trends Mol. Med. 22, 671–686 (2016).
Cuervo, A. M. & Macian, F. Autophagy and the immune function in aging. Curr. Opin. Immunol. 29, 97–104 (2014).
Phadwal, K. et al. A novel method for autophagy detection in primary cells: impaired levels of macroautophagy in immunosenescent T cells. Autophagy 8, 677–689 (2012).
Raz, Y. et al. Activation-induced autophagy is preserved in CD4+ T-cells in familial longevity. J Gerontol. A Biol. Sci. Med. Sci. 72, 1201–1206 (2017).
Alpert, A. et al. A clinically meaningful metric of immune age derived from high-dimensional longitudinal monitoring. Nat. Med. 25, 487–495 (2019).
Henson, S. M. et al. p38 signaling inhibits mTORC1-independent autophagy in senescent human CD8+ T cells. J. Clin. Invest. 124, 4004–4016 (2014).
Macian, F. Autophagy in T cell function and aging. Front. Cell Dev. Biol. 7, 213 (2019).
Wu, T. et al. The TCF1–Bcl6 axis counteracts type I interferon to repress exhaustion and maintain T cell stemness. Sci. Immunol. 1, eaai8593 (2016).
Han, S., Georgiev, P., Ringel, A.E., Sharpe, A.H. & Haigis, M.C. Age-associated remodeling of T cell immunity and metabolism. Cell Metab. 35, 36–55 (2023).
Palmer, D.C. et al. Internal checkpoint regulates T cell neoantigen reactivity and susceptibility to PD1 blockade. Med 3, 682–704.e8 (2022).
Neelapu, S. S. et al. Chimeric antigen receptor T-cell therapy - assessment and management of toxicities. Nat. Rev. Clin. Oncol. 15, 47–62 (2018).
Waldman, A. D., Fritz, J. M. & Lenardo, M. J. A guide to cancer immunotherapy: from T cell basic science to clinical practice. Nat. Rev. Immunol. 20, 651–668 (2020).
Mantovani, A., Allavena, P., Sica, A. & Balkwill, F. Cancer-related inflammation. Nature 454, 436–444 (2008).
Arthofer, E. et al. Genetic editing of CISH enhances T cell effector programs independently of immune checkpoint cell surface ligand expression. Preprint at bioRxiv https://doi.org/10.1101/2021.08.17.456714 (2021).
Baixauli, F. et al. Mitochondrial respiration controls lysosomal function during inflammatory T cell responses. Cell Metab. 22, 485–498 (2015).
Pence, B. D. et al. Relationship between systemic inflammation and delayed-type hypersensitivity response to Candida antigen in older adults. PLoS ONE 7, e36403 (2012).
Watanabe, R. et al. Pyruvate controls the checkpoint inhibitor PD-L1 and suppresses T cell immunity. J. Clin. Invest. 127, 2725–2738 (2017).
Teijaro, J. R. et al. Persistent LCMV infection is controlled by blockade of type I interferon signaling. Science 340, 207–211 (2013).
Wilson, E. B. et al. Blockade of chronic type I interferon signaling to control persistent LCMV infection. Science 340, 202–207 (2013).
McCausland, M. M. & Crotty, S. Quantitative PCR technique for detecting lymphocytic choriomeningitis virus in vivo. J. Virol. Methods 147, 167–176 (2008).
Steinke, F. C. et al. TCF-1 and LEF-1 act upstream of Th-POK to promote the CD4+ T cell fate and interact with Runx3 to silence Cd4 in CD8+ T cells. Nat. Immunol. 15, 646–656 (2014).
Acknowledgements
We thank the Mayo Clinic Microscopy and Cell Analysis Core for assistance with flow cytometry (C.A. Moe), cell sorting (Y.H. Han) and TEM (S. Gamb) and R. Ahmed (Emory University) for providing SMARTA mice and LCMV Armstrong. This work was supported by National Institutes of Health grants R01 AR042527, R01 HL117913, R01 AI108906 and R01 HL142068 to C.M.W. and R01 AI108891, R01 AG045779, U19 AI057266 and R01 AI129191 to J.J.G. I.S. was supported by T32AG049672 and by a Glenn Foundation for Medical Research Postdoctoral Fellow. The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Author information
Authors and Affiliations
Contributions
J.J., Y.M., C.M.W. and J.J.G. designed the study. J.J., Y.M., H.Z., I.S., C.W. and Q.X. performed the experiments. R.J. performed the computational analysis. J.J., Y.M., C.M.W. and J.J.G. analyzed and interpreted the data. J.J., Y.M. and J.J.G. wrote the manuscript with all authors providing feedback.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Aging thanks Maria Mittelbrunn, Vishwa Dixit, Pedro Rodrigues, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 TCF1 prevents mtDNA release by promoting lysosomal activity.
a, b, Naïve CD4+ T cells from older 7 females and 7 males were activated for 3 days (a). Cells from 3 young adults were activated in the presence or absence of 10 μM CQ or 5 nM BafA1 (b). mtDNA levels in the culture supernatants were determined. c, Lysosomal acidification was determined by flow cytometry-based analysis of LysoSensor in day-3-stimulated naïve CD4+ T cells comparing 8 young and 8 older individuals. d, Naïve CD4+ T cells from 3 older individuals were transfected with control or CISH siRNA and activated for 3 days. Reduced CISH protein expression after silencing was confirmed by immunoblotting. e, CISH transcripts in naïve CD4+ T cells and naïve CD8+ T cells stimulated for 3 days, in naïve B cells and CD14+ monocytes stimulated for 5 days comparing 6 young and 6 older individuals (left); mtDNA levels in culture supernatants of day-3-activated naïve CD 8+ T cells from 6 older individuals, transfected with siCtrl or siCISH (right). f, TCF protein levels in naïve CD4+ T cells at indicated time points after stimulation comparing 4 young and 4 older individuals. g, Naïve CD4+ T cells from 4 young individuals were transfected with control or TCF7 siRNA and activated for 3 days. Lysosomal acidification (left) and proteolytic activities (right) were determined by flow cytometry-based analysis of LysoSensor and DQ-BSA-treated cells, respectively. h, i, Naïve CD4+ T cells from older individuals were activated for 3 days in the presence of either vehicle or 100 nM of CHIR99021. Lysosomal acidification, proteolytic activities (n = 4) (h) and supernatant mtDNA levels (n = 3) (i) were determined. j, TCF1 and CISH protein in day-3-stimulated naïve CD4+ T cells from 3 older individuals after transfection with pCMV6 control vector or TCF7-expressing vector. Data are presented as mean ± s.e.m. Comparison by two-tailed unpaired t-test (a, c, e and f) or two-tailed paired t-test (b, d, e and g–j). *P < 0.05, **P < 0.01; NS, not significant.
Extended Data Fig. 2 Silencing of CISH and ATP6V1A expression in day 3-activated naïve CD4+ T cells.
a, Naïve CD4+ T cells were transfected with control or CISH siRNA and activated for 3 days (n = 3). Lysosomal acidification was determined by flow cytometry-based analysis of LysoSensor-treated cells. b, Naïve CD4+ T cells from a young and an older individual were co-stained with anti-CISH (green) and anti-ATP6V1A Ab (red). Confocal images representative of three independent experiments are shown. c, Proteasome activity in day 3-activated naïve CD4+ T cells from 6 young and 6 older individuals. d, Naïve CD4+ T cells from 3 young individuals were transfected with control or ATP6V1A siRNA and activated for 3 days. Reduced ATP6V1A protein expression after silencing was confirmed by Western blotting. Data are presented as mean ± s.e.m. Comparison by two-tailed paired t-test (a and d) or two-tailed unpaired t-test (c). *P < 0.05; NS, not significant.
Extended Data Fig. 3 CISH silencing does not affect the number of lysosomes per cell.
Quantitative assessment of lysosome numbers in day 3-activated naïve CD4+ T cells from 5 old individuals after CISH silencing. Image acquisition and analysis were performed by an examiner blinded to the nature of the specimen. Fifteen sections were analyzed for each donor, and mean number per section for each donor is shown NS, not significant. Data are presented as mean ± s.e.m. Comparison by two-tailed unpaired t-test.
Extended Data Fig. 4 Effect of CISH on mitochondria in activated T cells from older adults.
a, COX IV protein expression in day 3-stimulated naïve CD4+ T cells comparing 5 young and 5 older healthy individuals. b, Gating strategy for TMRMhiMtGhi, TMRMloMtGlo and TMRMloMtGhi. TMRMlo cells had reduced cell size (low FSC) and reduced density (low SSC) distinct from apoptotic cells. c, Frequencies of the TMRMloMtGlo population in day 3-activated naïve CD4+ T cells from 4 young and 4 older individuals after indicated gene silencing. Data are presented as mean ± s.e.m. Comparison by two-tailed unpaired t-test (a), one-way ANOVA followed by Tukey’s multiple comparison test (c). *P < 0.05, **P < 0.01 and ***P < 0.001; NS, not significant.
Extended Data Fig. 5 Confocal image analysis of the effect of CISH on mitochondria and MVBs.
a, b, Naïve CD4+ T cells from a young and an older individual were transfected with indicated siRNA and activated for 3 days. Cells were co-stained with anti-CD63 (as MVB marker) and with anti-LC3B (as autophagosome marker) (a) or anti-COX IV (as mitochondria marker) (b). Confocal images representative of two independent experiments are shown. Scale bars, 1 μm.
Extended Data Fig. 6 CISH-dependent amphisome-rich T cells from older individuals do not show evidence of apoptosis.
a, Naïve CD4+ T cells were transfected with control or indicated siRNA and activated for 3 days (n = 4). Cell apoptosis was examined by staining with Annexin V and 7-AAD. Statistical significance by two-tailed unpaired t-test. NS, not significant. b, Day3-stimulated naïve CD4+ T cells were left untreated or treated with 1 μM Staurosporine for 24 h. Scale bar, 1 μm. Cells were then processed for TEM. Data are presented as mean ± s.e.m.
Extended Data Fig. 7 TCF7 silencing promotes intracellular accumulation of amphisomes.
Naïve CD4+ T cells from young adults were transfected with control or TCF7 siRNA and activated for 3 days. Cells were then harvested and processed for TEM. Representative 30 TEM images (left) and quantitative plots of amphisome numbers in day 3-activated naïve CD4+ T cells from two young individuals after indicated transfection. Data collection and analysis were conducted in a blinded manner. Fifteen cells were analyzed for each donor. Scale bar, 1 μm. Data are presented as mean ± s.e.m. Statistical significance by two-tailed unpaired t-test. ****P < 0.0001.
Extended Data Fig. 8 Effect of CISH on T cell responses in vivo.
a–d, SMARTA cells were transduced and adoptively transferred followed by LCMV infection as described in Fig. 8. Viral RNA levels in the spleen were determined on day 6, n = 10 (a). Frequencies of SMARTA cells with indicated phenotypes in CD4+ donor cells (b–d) on day 6 after LCMV infection (n = 6). e, Mice were reconstituted with shCish or shCtrl retrovirally transduced naïve OT-II CD4+ T cells (n = 3). Frequencies and absolute numbers of TFH and non-TFH OT-II cells in the spleen on day 8 after OVA immunization. Data are presented as mean ± s.e.m. Comparison by two-tailed unpaired t-test (a–e). *P < 0.05; NS, not significant.
Extended Data Fig. 9 Confirmatory studies with a distinct shRNA to silence Cish.
a-d, 1 ×105 Cish shRNA#2 (5′-AAAACAAGTGTTAGAACACAA-3′) or control shRNA retrovirally transduced Amcyan+ naïve SMARTA CD4+ T cells were adoptively transferred into CD45.2+ naïve recipients followed by LCMV infection. a, CISH and ATP6V1A protein expression in transduced cells before adoptive transfer. b, Serum mtDNA copies at day 6 after LCMV infection. c, Serum proinflammatory cytokines levels on day 6 after LCMV infection. d, Gene expression of key inflammatory markers in splenocytes on day 6 after infection. Data are from one experiment with 3–4 mice per group. Data are presented as mean ± s.e.m. Statistical significance by one-way ANOVA followed by Tukey’s multiple comparison test (b and d) or two-tailed unpaired t-test (c). *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001; NS, not significant.
Extended Data Fig. 10
Gating strategy for TFH and non-TFH Amcyan+, successfully transduced cells in the spleen.
Supplementary information
Supplementary Table 1
Oligonucleotide primer sets used in this study.
Source data
Source Data All Figures
Statistical source data.
Source Data Fig. 1
Unprocessed western blots.
Source Data Fig. 2
Unprocessed western blots.
Source Data Fig. 3
Unprocessed western blots.
Source Data Fig. 8
Unprocessed western blots.
Source Data Extended Data Fig. 1
Unprocessed western blots.
Source Data Extended Data Fig. 2
Unprocessed western blots.
Source Data Extended Data Fig. 4
Unprocessed western blots.
Source Data Extended Data Fig. 9
nprocessed western blots.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Jin, J., Mu, Y., Zhang, H. et al. CISH impairs lysosomal function in activated T cells resulting in mitochondrial DNA release and inflammaging. Nat Aging 3, 600–616 (2023). https://doi.org/10.1038/s43587-023-00399-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s43587-023-00399-w
This article is cited by
-
Old T cells pollute with mito-litter
Nature Aging (2023)
