
Abstract
Lung adenocarcinoma is still cancer with the highest mortality. Hypoxia and immunity play an essential role in the occurrence and development of tumors. Therefore, this study is mainly to find new early diagnosis and prognosis markers and explore the relationship among the markers and immunity and hypoxia, to improve the prognosis of patients. Firstly, based on the clinical database in TCGA, we determined the most critical clinicopathological parameters affecting the prognosis of patients through a variety of analysis methods. According to pathological parameters, logistic most minor absolute contraction selection operator (lasso), univariate and multivariate regression analysis, the risk genes related to early prognosis were screened, and the risk model was established. Then, in different risk groups, GSEA and CIBERSORT algorithms were used to analyze the distribution and enrichment of the immune cells and hypoxia, to study the effects of early prognostic indicators on hypoxia and immunity. At the same time, we analyzed the different levels of risk genes in normal cells (BSEA-2B) and tumor cells (H1299, A549, PC9, and H1975). Finally, A549 and PC9 cells were induced by CoCl2 to establish a hypoxic environment, and the correlation between risk genes and HIF1A was analyzed. The risk model based on risk genes (CYP4B1, KRT6A, and FAM83A) was accurate and stable for the prognosis of patients. It is closely related to immunity and hypoxia. In BSEA-2B cells, the mRNA and protein expression of CYP4B1 was higher, while the expression of KRT6A and FAM83A was lower. Finally, we found that FAM83A and HIF1A showed a significant positive correlation when A549 and PC9 cells were exposed to hypoxia. The discovery of early diagnostic markers related to immunity, hypoxia, and prognosis, provides a new idea for early screening and prognostic treatment of lung adenocarcinoma.
Similar content being viewed by others
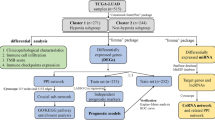

Introduction
Lung cancer is a disease with the highest mortality among all cancers, and non-small cell lung cancer accounts for about 85% of lung cancer1. Lung adenocarcinoma is the most common histological category of non-small cell lung cancer; It is rising among younger men and women2,3. Although surgery and combination therapy have been improving in recent years, the five-year survival rate of patients is still less than 20%4,5,6,7,8. Through a variety of modern methods, the prognosis of patients can be diagnosed and analyzed. Therefore, the TMN Stage has a higher five-year survival rate than other Stages9. Therefore, finding new early molecular prognostic markers can quickly and accurately determine the Stage of TMN, which is a very profitable strategy for patient treatment. Identifying the PD-1/PD-L1 immune checkpoint pathway as a therapeutic target for inducing an immune response has shown excellent prospects to respond to tumor cells10. However, this treatment is effective only for some (20–30%) patients11,12,13,14. At the same time, in most solid tumors, hypoxia is a crucial factor to promote the survival and adaptation of tumor cells and help cancer cells progress15. Therefore, Therefore, the development of new early prognostic markers related to immunity and hypoxia may improve the prognosis of patients with lung adenocarcinoma.
In this study, early diagnostic markers were screened using the gene transcriptome of the regular group and TMN Stage and the protein database of CPTAC, respectively. Then, risk genes were filtered from the early diagnosis and prognostic markers (edgps), using various statistical methods. At the same time, we also discussed the relationship between the risk model and immunity, hypoxia, mutation, and clinical prognosis. Finally, we studied the distribution difference of risk genes between tumor cells and normal cells and the correlation between HIF1A and risk genes in a hypoxia environment. In conclusion, this series of findings will provide ideas and strategies for patients' prognosis and early diagnosis.
Materials and methods
Data source
We downloaded the data sets of lung adenocarcinoma-related proteins and genes, including Cancer Genome Atlas (TCGA), Gene Expression Omnibus (GEO), and clinical proteomic Tumor Analysis Consortium. The gene transcription RNA-seq data and clinical information of 59 normal and 535 tumor patients, were downloaded from the TCGA database. The GEO (GSE26939) and CPTAC datasets contained 113 columns of clinical patient information, 102 normal, and 109 tumor proteomic data, respectively.
Screening of early diagnostic genes
We divided the RNA-seq data obtained from TCGA into two groups, including Stage I-normal and Stage I-Stage II-IV, which were analyzed for differential expression by “limma” package in R language; the screening criteria was |logFC|> 1 and 0.5, FDR < 0.5, respectively. Then, differential protein data was obtained through the normal and tumor groups of CPTAC and limma in R language; the screening criteria were |logFC |> 0.5, p value < 0.05. Three groups of differentially expressed genes and proteins were intersected to obtain early diagnostic genes (edgs).
Screening early diagnosis and prognosis genes and establishing early prognosis risk model
Firstly, after screening and dimensionality reduction of the early diagnosis and prognosis gene (edgps) through univariate and lasso Cox regression analysis, risk genes can be obtained. Then, after multivariate Cox regression analysis, the risk coefficient of risk genes (rgs) can be calculated to establish the early prognostic risk model (edgpsl). Finally, the calculation formula of the risk coefficient is as follows: risk score = (Exprgs1 × Coefrgs1) + (Exprgs2 × Coefrgs2) + … + (ExprgsN × CoefrgsN).
Survival difference and prognosis evaluation of high and low-risk groups
Survival analysis and prognosis evaluation were performed for high-risk and low-risk groups through the Kaplan Meyer method and ROC curve analysis.
Immune infiltrates analysis
TIMER is an online website that can evaluate the Infiltration degree of immune cells to various tumors16. CIBERSOFT can use a deconvolution-based algorithm to calculate the abundance of 22 immune cells in different tissues17. The data of LUAD related immune cell infiltrates score in TCGA are mainly downloaded from the CIBERSOFT algorithm on the TIMER website.
Functional enrichment analysis
David 6.8 bioinformatics resources and gene set enrichment analysis (GSEA) were used to perform GO and KEGG function enrichment analysis, respectively. The screening criteria of the GO pathway were p-value < 0.05, KEGG pathways with the following criteria were regarded as nominal p-value < 0.05.
Construction and evaluation of a predictive nomogram
Univariate and multivariate Cox regression analysis selected the most significant prognostic factors independently. It established a nomogram to predict the survival probability of one to three years, and the calibration value can be used to predict the prediction accuracy of the nomogram.
Cell source and culture environment
Lung adenocarcinoma cell lines were mainly purchased from the cell bank of Chinese Academy of Sciences (Shanghai, China), and cultured in RPMI-1640 medium (Gibco, Gaithersburg, MD, USA) with 10% fetal bovine serum (FBS) at 37 °C and 5% CO2.
Simulated cell hypoxia environment
The cells were planted overnight in a six well plate with a cell density of about 60–70%. The next day, 200 µ mol CoCl2 was added to each well. After 24–36 h, the protein was extracted and WB experiment was carried out. In the cell culture system, cobalt chloride (CoCl2) is a substance that induces cell hypoxia. CoCl2 inhibits the catalysis of proline hydroxylase so that the cells are in a state similar to hypoxia18,19,20.
Western blotting and RT qPCR experiment
The cells were cultured for 24 h, 1 ml Trizol was added, fully mixed and dissolved, operated according to the kit, and analyzed by RT-PCR. The cells were treated with complete protease inhibitor and Ripa lysate to extract protein. The protein concentration was obtained through the protein quantitative kit. Then, the electrophoresis instrument was modulated and stabilized at 70 V and 110 V for 90 and 60 min respectively, and the target protein was separated. Then, the steady flow 220A was modulated and the membrane was turned for 120 min. Then, the whole membrane was sealed with rapid sealing solution for 30 min. The ratio of working solution of primary antibody and secondary antibody was 1:1000 and 1:3000 respectively. Similarly, The incubation time of the target PVDF membrane is 12 h at 4 °C, and 2H at room temperature respectively. In the middle, the membrane needs to be washed three times for 10 min each time. Due to the small scope of cutting the PVDF membrane, the target antibody may be located at the boundary, but it has little effect on the overall result.
Statistical analysis
The “survivalroc” package of R (version 4.1.2) was used to analyze the ROC curve, and the “survivminer” and “glmnet” were combined for univariate, multivariate, lasso cox regression and survival difference analysis. The “RMS” package was used to draw the nomogram and calibration diagram, and then the “limma” package was used to analyze the difference of gene expression data. Other R packages that draw graphs of biological information related differences and correlations, including “ggpubr”, “pheatmap”, “ggplot2”, “ggpubr”, “ggextra” and “corrplot” packages in R software. Graphpad prism 8 is used to analyze and draw WB and PCR results.
Ethics approval
The data used in this study were obtained from publicly available datasets, such as the GEO database (HTTPS:// www. NCBI. nlm. nih. gov/ geo), and The Cancer Genome Atlas (HTTPS:// portal.GDC. cancer. gov). The KEGG and go pathway analysis used was from David database (https://david.ncifcrf.gov/summary.jsp).
Result
Determine the most significant independent prognostic clinical factors
In univariate and multivariate analysis, we found that only the p values of TMN Stage were all less than 0.05 (Fig. 1A, B). The AUC values of various clinicopathological factors (age, gender, TMN stage, T stage, M stage, and N stage) were 0.471, 0.558, 0.671, 0.576, 0.496, and 0.636, respectively (Fig. 1C), and the AUC value of TMN Stage was the highest. They are divided into two groups according to the TMN StageI and TMN StageII-IV. The survival rates of the two groups were significantly different, the prognosis of TMN StageII-IV was worse than that of other groups (p < 0.001) (Fig. 1D).

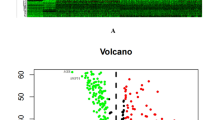
Screening early diagnostic genes related to TMNStage. (A, B), Forest plots showed univariate (A) and multivariate Cox (B) analysis of clinical characteristics. (C) ROC curve plots of clinical features. (D) Survival analysis between stage I and stage II-IV groups. (E, F) Heat, and (F) volcano map of differentially expressed genes between StageI and normal groups from the TCGA database. (G) Based on the Cptac database, the heat map showed the differentially expressed proteins between the normal and tumor group. (H) Based on the TCGA database, Heatmap showed the differentially expressed genes between StageI and II-IV. (I) Wayne’s diagram indicates that early diagnosis genes are screened via the overlaps of differential genes of the three groups.
Screening early diagnostic genes
Based on the normal and Stage groups in TCGA data, we used the Wilcoxon test method in R language to screen 7333 significant genes (Fig. 1E,F), and then differentially expressed between StageI and StageII-IV groups to obtain 296 significant genes (Fig. 1H). We also used the limma package in R language to analyze the differences between normal and tumor groups in the CPTAC database, To obtain 2234 significant genes (Fig. 1G). Finally, 38 early diagnosis genes (edgs) were obtained by the intersection of the above three groups of significant genes (Fig. 1I).
Screening early diagnosis and prognosis genes and establishing an early prognosis model
Firstly, 24 prognostic related edgs (edgps) (Fig. 2A) were screened by univariate Cox regression analysis, then, three early diagnostic markers (risk genes) with the greatest impact on prognosis edgps (Fig. 2B–D) were screened and calculated as risk scores based on lasso Cox regression dimensionality reduction and multivariate Cox regression analysis, to establish early prognostic risk model (edgpsl)16. The calculation formula of risk score is as follows: risk score = (−0.09 × Expression CYP4B1) + (0.1238 × Expression value of KRT6A) + (0.1022 × Expression value of FAM83A). Through the “survival” package in R language, the best threshold of clinical prognosis is selected from the risk score, which is used as the basis for the high-risk and low-risk groups. Through KM survival analysis, the study found that the prognosis of high-risk groups is very low, compared with the low-risk group in TCGA (Fig. 2E) and GEO (Fig. 2G) data sets. The AUC value of the three-year prognosis model of the two data sets (Fig. 2F,H) (TCGA: IYAUC = 0.711, 2YAUC = 0.682, 3YAUC = 0.680, GEO: IYAUC = 0.785, 2YAUC = 0.672, 3YAUC = 0.639) were analyzed based on the ROC curve in R soft.

Establish and verify the early diagnosis and prognosis model. (A) Univariate Cox survival analysis was performed for the early diagnosis genes. (B) A 1000-fold cross-validation for tuning parameter selection in the least absolute shrinkage and selection operator (LASSO) model. (C) LASSO coefficient profiles of the 24 early diagnosis and prognosis-related genes. (D) Multivariate Cox regression of three risk genes. E, G, KM survival analysis was performed to determine survival differences between different analysis groups, from TCGA and GEO (G) data sets. (F, H) The ROC curve revealed the AUC value of the prognostic model in the TCGA (F) and GEO (H) cohort.
Correlation between risk score and clinical features
We found that the risk scores were mainly distributed in the T2-4 Stage (Fig. 3D), N1-3 Stage (Fig. 3F), and TMN StageII-IV (Fig. 3C). However, there were no significant differences in the distribution of risk scores among age (Fig. 3A), gender (Fig. 3B), and M groups (Fig. 3E), compared with other clinical Stages.

The relationships between risk score and multiple clinicopathological parameters. (A) Age. (B) Gender. (C) TMNStage. (D) Tumor size (T). (E) Distant metastasis (M). (F) Lymph node status (N).
Distribution of immune cell infiltration under risk model
Through this study, we found that 10 immune cells were closely related to the risk score (Fig. 4B), including mast cell activated (cor = −0.34360408), T cell CD4 + memory resetting (cor = −0.226), Myeloid dendritic cell resting (cor = −0.185), B cell memory (cor = −0.174), Monocyte (cor = −0.16), Neutrophil (cor = 0.218), Mast cell resting (cor = 0.23), Macrophage M0 (cor = 0.239), NK cell resting(cor = 0.12) and Macrophage M1 (cor = 0.136). Then, by analyzing the difference of immune cell composition between high and low-risk groups (Fig. 4A), we found that NK cell resting Macrophage M1, Neutrophil, Mast cell resting, macrophage M0 and T cell CD4 + memory activated were mainly distributed in the high-risk group, while the immune cells mainly distributed in the low-risk group were mast cell activated, T cell CD4 + memory resting, myeloid dendritic cell resting, B cell memory, and Monocyte. Finally, compared with the low-risk group, the expression of immune-related genes programmed death-ligand 1 (PD-L1) and programmed cell death protein 1(PD1) were higher in the high-risk group (Fig. 4C). In the low-risk group, the expression levels of interleukin-4 (IL-4) and surface molecule CD20 (CD20) were higher than those in the high-risk group (Fig. 4D).

Compositions of infiltrated immune cells between different risk groups in TCGA-LUAD cohort. (A) The box plot shows the ratio differentiation of 22 kinds of immune cells between LUAD tumor samples with the high/low risk groups. (B) The radar chart shows the correlation between risk score and immune cell score. (C, D) Violin diagram shows the distribution level of immune related genes in high and low expression groups.
Hypoxia effects in the risk model
Based on the HALLMARK data set in the GSEA tool, we analyzed the functional enrichment of high and low-risk groups under the risk model. The high-risk group was mainly enriched in hypoxia, glycolysis, and PI3K-Akt-mTOR signal pathway (Fig. 5A–C). The mutation load of the high-risk group was higher than that of the low-risk group (Fig. 5D). At the same time, hypoxia-inducible factor A (HIF1A) and lactate dehydrogenase A (LDHA) were higher in the high-risk group than in other groups (Fig. 5E). In the high HIF1A group, the expression levels of FAM83a and KRT6A were higher, while CYP4B1 was the opposite (Fig. 5F). In addition, FAM83A and KRT6A were positively correlated with HIF1A, while CYP4B1 was contrary (Fig. 5G).

Effects of hypoxia in high and low-risk groups. (A–C), GSEA analysis showed that the high-risk group was enriched in hypoxia (A), glycolysis (B), and PI3K/Akt/mTOR (C) signaling pathway. (D) Tumor mutation load in different risk groups. (E) Expression levels of hypoxia-inducible factor HIF1A and hypoxia regulatory factor LDHA. (F) Distribution level of risk genes in high and low HIF1A expression groups. (G) Correlation between risk genes and HIF1A.
Establish and verify nomogram based on TMN Stage and risk score
Univariate and multivariate Cox regression analysis of multiple clinical factors and risk scores showed that TMN Stage and risk score had a significant impact on the prognosis of patients. The results of univariate and multivariate analysis showed that TMN Stage (UNIX: HR = 2.918, 95% Cl = 1.989–4.283, p < 0.001. Mutiox: HR = 4.187, 95% Cl = 1.627–10.776, p = 0.003) (Fig. 6A) and risk score (UNIX: HR = 1.682, 95% Cl = 1.354–2.689, p < 0.001. Mutiox: HR = 1.611, 95% Cl = 1.275–2.035, p < 0.001) (Fig. 6B). AUC values of various clinical factors and risk scores were analyzed in the "TIMER ROC" package in R language, among which TMN Stage (AUC = 0.669) and risk score (AUC = 0.711) is the highest (Fig. 6C). The nomogram was established based on TMN Stage and risk score to predict the three-year survival rate of patients (Fig. 6D), and the range of C index of 95% confidence interval was 0.6598 to 0.7292. The deviation correction line was consistent with the calibration curve, indicating that the nomogram had high prediction ability (Fig. 6E).

Establishment and evaluation of predictive nomogram in early diagnosis and prognosis model. (A, B), Forest map showed that independent clinical prognostic factors were obtained by univariate (A) and multivariate (B) Cox analysis. (C) The ROC curve exhibited the predictive performance of each independent predictive factor. (D) A nomogram for predicting OS in significantly independent prognostic factors. (E) Calibration plot of the nomogram for the probability of OS at 1, 2 and 3 year.
Functional enrichment analysis of KEGG and GO related to different risk groups
Based on the KEGG data set in GSEA, we found that the low-risk group is mainly enriched in α-Linolenic acid metabolism, arachidonic acid metabolism, and Linoleic acid metabolism (Fig. 7B). In contrast, high-risk groups were primarily increased in bladder cancer, cell cycle, replication of bladder, pancreatic cancer, renal cell carcinoma, and small cell carcinoma signal pathway (Fig. 7A). GO function enrichment analysis was conducted through the DAVID online website for significant genes in high and low-risk groups. The highly expressed genes in high-risk groups were mainly enriched in skin development, keratinocyte differentiation, epithelial cell differentiation, epidermis development, cornification, intermediate filing, intermediate filing cytoskeleton, keratin filing, and structural constitution cytoskeleton, certified envelope, and calcium-dependent protein binding (Fig. 7C). The significant genes in the low expression group were mainly enriched in the receptor-mediated endocytosissteroid metabolic process, alcohol metabolic process, terpenoid metabolic process, antimicrobial humoral response, blood microparticle, endocytic vesicle, secretory granule lumen, cytoplasmic vesicle lumen, and vesicle lumen (Fig. 7D).

Functional enrichment analysis of KEGG and GO in high and low risk groups. (A, B), Gene set enrichment analysis (GSEA) results show the enriched KEGG pathways in the high (A) and low (B) risk groups. (C, D), Bubbles (C) and bar (D) plots show enriched go pathways based on up-regulated gene analysis in high-risk and low-risk groups, respectively.
Univariate Cox regression was used to analyze the survival results of OS, DSS, DFI, and PFI related to early prognostic genes
For the univariate cox analysis of early prognostic genes (FAM83A, KRT6A, and CYP4B1) related to OS, DSS, DFI, and PFI of 32 tumors, we selected the top one to three with the lowest p-value in the analysis of tumor prognosis. Firstly, in the analysis of overall survival (OS), FAM83A (p = 2.06e-07), KRT6A (p = 1.22e-07), and CYP4B1 (p = 6.72e-05) had the most significant effect on the overall survival of patients with lung adenocarcinoma. Patients with high expression of FAM83A (HR = 1.283,95% CI = 1.168–1.410) and KRT6A (HR = 1.156, 95% CI = 1.097–1.223) had lower survival, while CYP4B1 (HR = 0.866,95% CI = 0.806–0.929) had the opposite effect. In the prognostic analysis of disease-free interval (DFI), CYP4B1 (p = 0.003) and FAM83A (p = 0.003) had the highest effect on the disease-free progression of lung adenocarcinoma, while KRT6A (p = 0.046) had a higher effect on BRCA. High expression of KRT6A (HR = 1.108, 95%CI = 1.004–1.223) and FAM83A (HR = 1.227, 95%CI = 1.074–1.402) had a poor prognostic progression of BRCA and lung adenocarcinoma, while CYP4B1 had the opposite effect on lung adenocarcinoma (HR = 0.860, 95%CI = 0.778–0.950). In the prognostic analysis of disease-specific survival (DSS), CYP4B1 (p = 0.000249), FAM83A (p = 5.69E-05), and KRT6A (p = 0.001235) had the highest effect on the disease-specific survival of lung adenocarcinoma patients. High expression of KRT6A (HR = 1.125, 95%CI = 1.047–1.207) and FAM83A (HR = 1.288, 95%CI = 1.138–1.457) was associated with poor DSS of lung adenocarcinoma, while CYP4B1 had the opposite effect on lung adenocarcinoma. In the prognostic analysis of progression-free interval (PFI), CYP4B1 (p = 6.40e-05) and KRT6A (p = 0.0111) had the highest impact on PFI in patients with lung adenocarcinoma, and FAM83A had a higher impact on PFI in KIRC (p = 0.0002052). High expression of KRT6A (HR = 1.075, 95% CI = 1.017–1.137) and FAM83A (HR = 2.436, 95% CI = 1.522–3.898) were associated with poor PFI of KIRC, while CYP4B1 (HR = 0.874,95% CI = 0.819–0.934) had the opposite effect on lung adenocarcinoma (Tables 1, 2, 3).
Compared with normal and StageI groups, KRT6A and FAM83A were higher in StageII-IV groups, while CYP4B1 expression levels were opposite (Fig. 8A–C). Similarly, in the analysis of the CPTAC dataset in the UALCAN database, the protein expression levels of KRT6A and FAM83A were higher in the tumor group, while CYP4B1 was the opposite (Fig. 8D–F). The mRNA and protein expression levels of FAM83A and KRT6A in tumor cells A549, PC9, and H1975 were higher than those of normal cells BSE-2B, while the mRNA and protein expression level of CYP4B1 in normal cells BSE-2B was higher than that of tumor cells H1299, PC9, A549 and H1975 (Fig. 9A–E) (*p < 0.05, **p < 0.01, ***p < 0.001).

Prognosis and expression results of early diagnosis and prognosis genes in lung adenocarcinoma and other tumors. (A–C), CYP4B1 (A), FAM83A (B), and KRT6A (C) were expressed in the normal group, stage I, and stage II-IV groups. (D–F), The expression levels of CYP4B1, FAM83A, and KRT6A in normal and tumor groups were analyzed by using the CPTAC data set in UNLCAN database.

Expression levels of risk genes in normal and tumor cells. (A–C), MRNA expression levels of KRT6A (A), FAM83A (B) and CYP4B1 (C) in normal and lung adenocarcinoma cells. (D, E), Protein expression level of risk genes.
Correlation between risk genes and HIF1A in a hypoxic environment
HIF1A increased significantly after CoCl2 induced A549 and PC9 cells. Meanwhile, in A549 cells, KRT6A and FAM83A increased with HIF1A. In PC9 cells, only FAM83A increased with HIF1A. The changing trend of CYP4B1 is not apparent (Fig. 10 A-B).

(A, B) CoCl2 induced hypoxia and normal environment, the expression level of risk genes (KRT6A, FAM83A and CYP4B1) and HIF1A.
Discussion
This study evaluated the prognosis and accuracy of various clinicopathological parameters through the univariate, multivariate Cox regression and ROC curve analysis. In different TMN Stage groups, there was a significant survival difference between Stage I and Stage II-IV groups; TMN Stage has the most noticeable impact on the prognosis of patients. In the past study, we also found that TMN Stage can predict the survival probability of patients9. At the same time, LUAD, as a fatal malignant tumor, can lead to the five-year survival rate of patients being still very low21. In conclusion, we can improve the prognosis of patients by grouping TMN Stage to obtain new potential early diagnostic and prognostic markers. Thirty-eight early diagnosis genes (edgs) were selected from three groups of genes with significant differences, including StageI v Normal, StageI v II-IV, and CPTAC. Then, based on the univariate, lasso, and multivariate Cox regression analysis, three risk genes were selected from edgps and calculated to establish a risk model. The three risk genes were CYP4B1, FAM83A, and KRT6A, respectively. As an extrahepatic form of cytochrome P450, CYP4B1 is mainly highly expressed in the lung and a small amount in other organs22. Decreased CYP4B1 is associated with poor prognosis in patients with bladder cancer23. Inhibit CYP4B1 can promote the occurrence of lung adenocarcinoma by preventing metabolism, enhancing DNA replication, and cell cycle activity; when the specific situation is unknown, it needs to be verified by experiments24. FAM83A is a member of an 8-member protein family. They have a highly conserved N-terminal domain with the same function and unknown function called the duf1669 domain25. FAM83A has been proved to promote the proliferation, invasion, stem cell-like characteristics, and drug resistance of lung cancer, breast cancer, and pancreatic cancer26,27,28,29,30,31. Keratin 6A (KRT6A) is a type II keratin involved in the epimerization of squamous epithelium32,33. Recent studies found that KRT6A plays a vital role in cell migration, especially keratinocyte migration. Downregulation of KRT6A expression can inhibit cell invasion and metastasis of nasopharyngeal carcinoma. In lung adenocarcinoma, a high KRT6A level is associated with poor prognosis and can promote the growth and metastasis of lung adenocarcinoma by inducing epithelial-mesenchymal transformation34,35,36,37,38.
Interestingly, using OS, DSS, DFI, and PFI related univariate Cox regression analysis in various tumors, we found that FAM83A, CYP4B1, and KRT6A had the most significant impact on the survival and progression of lung adenocarcinoma. In the normal tissue and TMN Stage group, FAM83A and KRT6A were the highest in the TMN Stage II-IV group, while CYP4B1 was the opposite. In normal and tumor cells, FAM83A and KRT6A were expressed higher in most tumor cells than in normal cells, while CYP4B1 was the opposite. In conclusion, the three risk genes significantly impact the early diagnosis and prognosis of patients with lung adenocarcinoma. The patients were divided into high and low-risk groups, according to the risk score in the early prognosis model. In the correlation analysis of multiple clinicopathological indexes and risk scores, we found that the risk scores in T2-4, N1-3, and StageII-IV groups were generally higher than those in other groups. Then, through univariate and multivariate cox regression analysis, we found that TMN Stage and risk score was the more significant independent prognostic factors. The two independent prognostic factors were used to establish a nomogram with good prediction ability and accuracy.
Using the immune score analyzed by the CIBERSORT method on the TIMER website, we found that NK cell resting, macrophage M1, neutrophil, mass cell resting, and macrophage M0 cells were distributed higher in a high-risk group, and the risk score was positively correlated with these immune cells. Mass cell activated, T cell CD4 + memory resting, myeloid dendritic cell resting, B cell memory, and monocyte cells were highly distributed in the low-risk group and negatively correlated with a risk score. Through David's online website's GO function enrichment analysis, the essential immune-related pathway in the low-risk group is a human antimicrobial response; B cell plays a vital role in humoral immunity39,40. Interleukin 4 (IL4) and B cell surface receptor CD20, which promote the proliferation and development of B cells, were highly expressed in the risk group41,42,43,44,45,46.
Based on the hallmark data set analysis in the GSEA tool, the high-risk group was mainly enriched in hypoxia, PI3K-Akt -mTOR, and the glycolysis signal pathway. At the same time, the HIF1A, LDHA, and mutation load are higher in the high-risk group than in the risk group. In previous studies, tumors in a hypoxic environment are more likely to lead to poor prognosis and mutations47,48. Under hypoxic conditions, HIF1A expression increases and induces downstream signal pathways, and PI3K -Akt-mTOR signaling pathway can promote tumor proliferation49,50,51,52,53. Glycolysis can enhance tumor proliferation, and it is a crucial enzyme to promote glycolytic activity54,55,56. Interestingly, HIF1A can also regulate glycolysis through the PI3K-Akt-mTOR signaling pathway57. Through the analysis of KEGG data set in the GSEA tool, the high-risk group is mainly enriched in the signal pathways related to cancer, such as boulder cage, cell cycle, DNA replication, glycolysis gluconeogenesis, pancreatic cage, renal cell carcinoma, and small cell lung. The low expression group was mainly enriched in alpha-linolenic acid metabolism, linoleic acid metabolism, and arachidonic acid metabolism pathway. In related learning, we found that alpha-linolenic acid and arachidonic acid metabolism can inhibit tumors and promote apoptosis58,59,60,61. Finally, through RT-PCR and WB, we found that the expression levels of tumor cells (A549, H1975, and A549) in BESA-2B cells were significantly higher than those in FAM83A and KRT6A, while CYP4B1 was the opposite. In addition, FAM83A and HIF1A are positively correlated, after CoCl2 induced hypoxia. This discovery has opened up new ideas for us.
In conclusion, based on various bioinformatics methods and cytological experiments, the screened risk genes can be used as potential early prognostic diagnostic markers of lung adenocarcinoma, which is also closely related to the development of immunity and hypoxia. This series of findings will provide a new idea for the comprehensive treatment of LUAD.
Data availability
Publicly available datasets were analyzed in this study; these can be found in the GEO database (https://www.NCBI.nlm.nih.gov/geo), and The Cancer Genome Atlas (https://portal.GDC.cancer.gov). The authors confirm that the data supporting the findings of this study are available within the article and its Supplementary materials. A statement that all methods and public data are implemented by relevant guidelines and regulations.
References
Torre, L. A. et al. Global cancer statistics, 2012. CA A Cancer J. Clin. 65, 87–108 (2015).
Hirsch, F. R. et al. Lung cancer: Current therapies and new targeted treatments. Lancet (London, England). 389, 299–311 (2017).
Mao, Y. et al. Epidemiology of lung cancer. Surg. Oncol. Clin. N. Am. 25, 439–445 (2016).
Chen, D. & Tan, Q. Comment on “robotic versus video-assisted lobectomy/segmentectomy for lung cancers-a meta-analysis”. Ann. Surg. 270, e147–e148 (2019).
Galvez, C. et al. Nonintubated uniportal VATS pulmonary anatomical resections. J. Vis. Surg. 3, 120 (2017).
Couñago, F. et al. Neoadjuvant treatment followed by surgery versus definitive chemoradiation in stage IIIA-N2 non-small-cell lung cancer: A multi-institutional study by the oncologic group for the study of lung cancer (Spanish Radiation Oncology Society). Lung cancer (Amsterdam, Netherlands). 118, 119–127 (2018).
Soria, J. C. et al. Osimertinib in untreated EGFR-mutated advanced non-small-cell lung cancer. N. Engl. J. Med. 378, 113–125 (2018).
Siegel, R. L. et al. Cancer statistics, 2018. CA A Cancer J. Clin. 68, 7–30 (2018).
Goldstraw, P. et al. The IASLC lung cancer staging project: Proposals for the revision of the TNM stage groupings in the forthcoming (seventh) edition of the TNM Classification of malignant tumours. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer. 2, 706–714 (2007).
Sharma, P. & Allison, J. P. The future of immune checkpoint therapy. Science (New York, NY). 348, 56–61 (2015).
Rittmeyer, A. et al. Atezolizumab versus docetaxel in patients with previously treated non-small-cell lung cancer (OAK): A phase 3, open-label, multicentre randomised controlled trial. Lancet (London, England). 389, 255–265 (2017).
Herbst, R. S. et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): A randomised controlled trial. Lancet (London, England). 387, 1540–1550 (2016).
Brahmer, J. et al. Nivolumab versus docetaxel in advanced squamous-cell non-small-cell lung cancer. N. Engl. J. Med. 373, 123–135 (2015).
Borghaei, H. et al. Nivolumab versus docetaxel in advanced nonsquamous non-small-cell lung cancer. N. Engl. J. Med. 373, 1627–1639 (2015).
Zhou, J. et al. Tumor hypoxia and cancer progression. Cancer Lett. 237, 10–21 (2006).
Li, T. et al. TIMER: A web server for comprehensive analysis of tumor-infiltrating immune cells. Can. Res. 77, e108–e110 (2017).
Newman, A. M. et al. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 12, 453–457 (2015).
Rovetta, F. et al. Cobalt triggers necrotic cell death and atrophy in skeletal C2C12 myotubes. Toxicol. Appl. Pharmacol. 271, 196–205 (2013).
Cervellati, F. et al. Hypoxia induces cell damage via oxidative stress in retinal epithelial cells. Free Radic. Res. 48, 303–312 (2014).
Yuan, Y. et al. Cobalt inhibits the interaction between hypoxia-inducible factor-alpha and von Hippel-Lindau protein by direct binding to hypoxia-inducible factor-alpha. J. Biol. Chem. 278, 15911–15916 (2003).
Denisenko, T. V. et al. Cell death-based treatment of lung adenocarcinoma. Cell Death Dis. 9, 117 (2018).
Wiek, C. et al. Identification of amino acid determinants in CYP4B1 for optimal catalytic processing of 4-ipomeanol. Biochem. J. 465, 103–114 (2015).
Lin, J. T. et al. Downregulation of the cytochrome P450 4B1 protein confers a poor prognostic factor in patients with urothelial carcinomas of upper urinary tracts and urinary bladder. APMIS Acta Pathologica, Microbiologica, et Immunologica Scandinavica 127, 170–180 (2019).
Liu, X. et al. CYP4B1 is a prognostic biomarker and potential therapeutic target in lung adenocarcinoma. PLoS ONE 16, e0247020 (2021).
Chen, S. et al. FAM83A is amplified and promotes cancer stem cell-like traits and chemoresistance in pancreatic cancer. Oncogenesis 6, e300 (2017).
Snijders, A. M. et al. FAM83 family oncogenes are broadly involved in human cancers: An integrative multi-omics approach. Mol. Oncol. 11, 167–179 (2017).
Richtmann, S. et al. FAM83A and FAM83B as prognostic biomarkers and potential new therapeutic targets in NSCLC. Cancers 11, 652 (2019).
Shi, R. et al. Long noncoding antisense RNA FAM83A-AS1 promotes lung cancer cell progression by increasing FAM83A. J. Cell. Biochem. 120, 10505–10512 (2019).
Lee, S. Y. et al. FAM83A confers EGFR-TKI resistance in breast cancer cells and in mice. J. Clin. Investig. 122, 3211–3220 (2012).
Grant, S. FAM83A and FAM83B: Candidate oncogenes and TKI resistance mediators. J. Clin. Investig. 122, 3048–3051 (2012).
Zhou, F. et al. FAM83A signaling induces epithelial-mesenchymal transition by the PI3K/AKT/Snail pathway in NSCLC. Aging 11, 6069–6088 (2019).
Fujii, T. et al. A preliminary transcriptome map of non-small cell lung cancer. Can. Res. 62, 3340–3346 (2002).
Chang, H. H. et al. A transcriptional network signature characterizes lung cancer subtypes. Cancer 117, 353–360 (2011).
Wang, F. et al. Keratin 6 regulates collective keratinocyte migration by altering cell-cell and cell-matrix adhesion. J. Cell Biol. 217, 4314–4330 (2018).
Chen, C. & Shan, H. Keratin 6A gene silencing suppresses cell invasion and metastasis of nasopharyngeal carcinoma via the β-catenin cascade. Mol. Med. Rep. 19, 3477–3484 (2019).
Shan, C. et al. 4-hydroxyphenylpyruvate dioxygenase promotes lung cancer growth via pentose phosphate pathway (PPP) flux mediated by LKB1-AMPK/HDAC10/G6PD axis. Cell Death Dis. 10, 525 (2019).
Xiao, J. et al. Eight potential biomarkers for distinguishing between lung adenocarcinoma and squamous cell carcinoma. Oncotarget 8, 71759–71771 (2017).
Yang, B. et al. KRT6A Promotes EMT and cancer stem cell transformation in lung adenocarcinoma. Technol. Cancer Res. Treat. 19, 1533033820921248 (2020).
Victora, G. D. & Nussenzweig, M. C. Germinal centers. Annu. Rev. Immunol. 30, 429–457 (2012).
Mesin, L. et al. Germinal center B Cell dynamics. Immunity 45, 471–482 (2016).
Howard, M. et al. Identification of a T cell-derived b cell growth factor distinct from interleukin 2. J. Exp. Med. 155, 914–923 (1982).
Isakson, P. C. et al. T cell-derived B cell differentiation factors effect on the isotype switch of murine B cells. J. Exp. Med. 155, 734–748 (1982).
Stashenko, P. et al. Characterization of a human B lymphocyte-specific antigen. J. Immunol. (Baltimore, Md. 1950) 125, 1678–1685 (1980).
Stashenko, P. et al. Expression of cell surface markers after human B lymphocyte activation. Proc. Natl. Acad. Sci. USA 78, 3848–3852 (1981).
Rosenthal, P. et al. Ontogeny of human hematopoietic cells: analysis utilizing monoclonal antibodies. J. Immunol. (Baltimore Md: 1950) 131, 232–237 (1983).
Nadler, L. M. et al. B4, a human B lymphocyte-associated antigen expressed on normal, mitogen-activated, and malignant B lymphocytes. J. Immunol. (Baltimore, Md 1950) 131, 244–250 (1983).
Bristow, R. G. & Hill, R. P. Hypoxia and metabolism Hypoxia, DNA repair and genetic instability. Nat. Rev. Cancer 8, 180–192 (2008).
Luoto, K. R. et al. Tumor hypoxia as a driving force in genetic instability. Genome Integr 4, 5 (2013).
Kaelin, W. G. Jr. & Ratcliffe, P. J. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol. cell. 30, 393–402 (2008).
Peet, D. J. et al. Oxygen-dependent asparagine hydroxylation. Methods Enzymol. 381, 467–487 (2004).
Niu, G. et al. Signal transducer and activator of transcription 3 is required for hypoxia-inducible factor-1alpha RNA expression in both tumor cells and tumor-associated myeloid cells. Mol. Cancer Res.: MCR. 6, 1099–1105 (2008).
Arafeh, R. & Samuels, Y. PIK3CA in cancer: The past 30 years. Semin. Cancer Biol. 59, 36–49 (2019).
Murugan, A. K. et al. Genetic deregulation of the PIK3CA oncogene in oral cancer. Cancer Lett. 338, 193–203 (2013).
Cantor, J. R. & Sabatini, D. M. Cancer cell metabolism: one hallmark, many faces. Cancer Discov. 2, 881–898 (2012).
Cui, J. et al. FOXM1 promotes the warburg effect and pancreatic cancer progression via transactivation of LDHA expression. Clin. Cancer Res.: An Off. J. Am. Assoc. Cancer Res. 20, 2595–2606 (2014).
Jiang, W. et al. FOXM1-LDHA signaling promoted gastric cancer glycolytic phenotype and progression. Int. J. Clin. Exp. Pathol. 8, 6756–6763 (2015).
Agani, F. & Jiang, B. H. Oxygen-independent regulation of HIF-1: Novel involvement of PI3K/AKT/mTOR pathway in cancer. Curr. Cancer Drug Targets 13, 245–251 (2013).
Wang, S. C. et al. α-Linolenic acid inhibits the migration of human triple-negative breast cancer cells by attenuating Twist1 expression and suppressing Twist1-mediated epithelial-mesenchymal transition. Biochem. Pharmacol. 180, 114152 (2020).
Klein, V. et al. Low alpha-linolenic acid content of adipose breast tissue is associated with an increased risk of breast cancer. Eur. J. Cancer Oxford, England 1990(36), 335–340 (2000).
Hannah, V. C. et al. Unsaturated fatty acids down-regulate srebp isoforms 1a and 1c by two mechanisms in HEK-293 cells. J. Biol. Chem. 276, 4365–4372 (2001).
Bae, S. et al. Arachidonic acid induces ER stress and apoptosis in HT-29 human colon cancer cells. Anim. Cells Syst. 24, 260–266 (2020).
Funding
This study was supported by grants from the Key Natural Science Project of Anhui Provincial Education Department (Nos. KJ2018A0221, KJ2020A0578 and KJ2021A0773) and National Innovation Program for College Students (202210367076), Anhui Provincial Undergraduate Innovative Training Program (Nos. S202010367008 and S202110367030).
Author information
Authors and Affiliations
Contributions
KS, CL, and JZ provide the idea and design of this article. Clinical data were collected and analyzed by KS, ZZ,YH and DW. KS and CLdrafted the first draft of the article and the drawing of charts. CL and JZ reviewed the revised paper. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sun, K., Zhang, Z., Wang, D. et al. Regulation of early diagnosis and prognostic markers of lung adenocarcinoma in immunity and hypoxia. Sci Rep 13, 6459 (2023). https://doi.org/10.1038/s41598-023-33404-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-33404-8
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
