
Abstract
Protective microbes have a major role in shaping host–pathogen interactions, but their relative importance in the structure of the host microbiota remains unclear. Here, we used a network approach to characterize the impact of a novel, experimentally evolved ‘protective microbial symbiont’ (Enterococcus faecalis) on the structure and predicted function of the natural microbiota of the model organism Caenorhabditis elegans. We used microbial network analysis to identify keystone taxa and describe the hierarchical placement of protective and non-protective symbionts in the microbiota. We found that early colonization with symbionts produce statistically significant changes in the structure of the community. Notably, only the protective E. faecalis became a keystone taxon in the nematode microbiota. Non-protective lineages of the same bacterial species remained comparatively unimportant to the community. Prediction of functional profiles in bacterial communities using PICRUSt2 showed that the presence of highly protective E. faecalis decreased the abundance of ergothioneine (EGT) biosynthesis pathway involved in the synthesis of the antioxidant molecule EGT, a potential public good. These data show that in addition to direct antagonism with virulent pathogens, keystone protective symbionts are linked to modified bacterial community structure and possible reductions in public goods, potentially driving decreased antioxidant defense. We suggest that this response could suppress infection via wholesale microbial community changes to further benefit the host. These findings extend the concept of protective symbionts beyond bodyguards to ecosystem engineers.
Similar content being viewed by others

Introduction
Animals have complex ecological interactions with their associated microbes1. Some of these micro-organisms are known as protective symbionts as they can defend their hosts against pathogen infection2,3. Whether protective symbionts were acquired by the host from the environment or inherited3, interactions with other colonizing microbes may occur within the host. The protective phenotype may emerge from host-microbe interactions4. Alternatively, direct interactions between the symbionts and pathogens can result in disease suppression via resource or interference competition5,6.
Whilst some protective symbionts are intracellular7, others may be embedded and interact with other host-microbiota components8,9. It has been previously shown that the protective effect can be maintained despite multi-species interactions within the microbiota, with the symbionts having minimal impact on native microbial diversity10. Whether protective symbionts can evolve to become important factors in the microbial community hierarchy is unclear. Some keystone taxa, with a major influence on microbiome structure and function at a particular space or time11, have been linked with host defense12. For example, keystone taxa are associated with protection against ‘bacterial wilt’, a common soil-borne disease caused by Ralstonia solanacearum in tobacco plants12. Other keystone species in the microbiota play key roles in the regulation of human gut metabolism13. The manipulation of keystone taxa is a promising approach to induce changes in the microbial community composition and functioning via taxa depletion or inoculation. Depletion of the keystone genera Escherichia-Shigella via host antibodies produced major shifts in the tick microbiota and affected tick feeding14,15. Moreover, inoculation of three Pseudomonas isolates, P. lurida FGD5-2, P. koreensis HCH2-3, and P. rhodesiae MTD4-1 displayed strong inhibitory effects on R. solanacearum12. Keystone taxa provide important services to the microbial community but can also participate in host defense.
The model organism Caenorhabditis elegans has a natural, resident microbiota1,16,17. A meta-analysis of C. elegans microbiota data shows that, regardless of the origin of the worms, whether field or laboratory isolates, C. elegans exhibit a core microbiota dominated by the bacterial families as Enterobacteriaceae, Pseudomonaceae, Xanthomonodaceae, Sphingobacteriaceae, Weeksellaceae and Flavobacteriaceae18. However, rare taxa, such as Acetobacteriaceae, were also consistently recovered from the C. elegans microbiota18. Acetobacteriaceae was identified as a possible keystone taxon associated with large populations of C. elegans proliferating in rotting apples1, which suggests a beneficial role of this keystone taxon on host physiology. Gut microbiota also contributes to C. elegans immunity and resistance to bacterial infections. King et al.19 demonstrated that co-adaptation between C. elegans and a novel, resident bacterium. Enterococcus faecalis, during infection by the pathogen Staphylococcus aureus can select for host-protection by E. faecalis.19. When C. elegans is colonised by evolved, protective E. faecalis, and subsequently exposed to a complex compost microbiota, there is minimal impact on the host microbiota diversity10.
Inference of microbial co-occurrence networks is a powerful tool for the study of microbial community structure and its dynamics20,21. Based on abundance data, obtained from high-throughput sequencing technologies, significant patterns of co-occurrence between microbial taxa can be detected and illustrated as networks20. Thus, nodes and links in a microbial network represent bacterial taxa and their respective co-occurrences22. Besides the prediction of bacterial associations, microbial co-occurrence network analysis offers the possibility to statistically identify keystones taxa. Although there is not a consensus on the metrics used for their identification, it is clear that keystone taxa are highly influential bacteria on the microbial community structure and functioning11,21,23. Apart from deciphering microbial community assembly patterns, 16S rRNA data can also be used for the prediction of metabolic function by matching taxonomic data to metabolic reference databases24,25. Thus, the exploration of microbial co-occurrence networks, identification of keystone taxa and functional prediction can give a comprehensive insight into the dynamics of the microbiota when a protective symbiont is introduced.
Here, we used a network approach to analyse 16S rRNA metabarcoding data from Dahan et al.10 and further characterized the impact of experimentally adapted, protective microbial lineages in the subsequent structure of the C. elegans-associated microbial community. Although broad diversity metrics (i.e., alpha/beta diversity) were unchanged10, we hypothesized that the introduction of highly protective lineages could alter microbe-microbe or microbe-host interactions and yield differences in overall community function that cannot be detected by taxonomy alone. We found that newly introduced microbes with strong protective phenotypes can become keystone taxa. Our results suggest that these symbionts play important roles in microbial community structure, as well as in host health. Within the text, “microbiome” refers to the microorganisms and their metagenome whereas “microbiota” refers only to the microbes themselves.
Methods
Original 16S rRNA datasets
In the present study, we used available 16S rRNA amplicon sequence datasets10. In the original study, Dahan et al.10 tested the impact of different bacterial strains on the microbiota of C. elegans. To this aim, E. faecalis OG1RF, originally isolated from the human gastrointestinal tract, was used as the ‘ancestral strain’ from which two lineages were derived by serial passages (non-protective and protective, Fig. 1A). The ‘non-protective E. faecalis’ evolved from ancestral E. faecalis in the absence of pathogen coinfection and did not confer protection to C. elegans. The ‘protective E. faecalis’ evolved from ancestral E. faecalis in the presence of a pathogenic Staphylococcus aureaus strain, and conferred protection against pathogen infection19 (Fig. 1A).

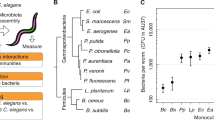
Graphical summary of the origin of different symbionts exposed to C. elegans. (Top panel) E. faecalis derived from a single ancestor clone, E. faecalis OG1RF, was used for an experimental evolution in C. elegans over 15 generations. In the experiment done by King et al.19, C. elegans was exposed to E. faecalis in the absence (green panel) or presence (pink panel) of a fixed, non-evolving pathogen S. aureus. Then, bacteria-killed nematodes were picked and crushed to release the pathogens from inside the carcass and the suspension was streaked on selective media to isolate populations of E. faecalis. Subsequently, 15 colonies were selected to make a lawn for the next exposure generation. From the single evolution, a non-protective E. faecalis was obtained while from the co-colonisation evolution, the protective E. faecalis strain was obtained. (Middle panel) The different strains of E. faecalis were then used for the exposition to C. elegans, in an independent experiment done by Dahan et al.10, alongside with P. mendocina, known to be protective against P. aeruginosa. E. coli OP50 was used as food control. (Bottom panel) 16s rRNA gene sequencing from C. elegans exposed to different symbionts were used for network analysis, keystone taxa identification and metagenomic prediction in the present study.
In addition to using the ancestral E. faecalis, non-protective E. faecalis and protective E. faecalis, Dahan et al.10 tested the impact of Pseudomonas mendocina of the microbiota of C. elegans (Fig. 1B). Pseudomonas mendocina was selected based on its capability to elicit host defenses against the pathogen Pseudomonas aeruginosa and was originally isolated from soil26. Escherichia coli OP50 was used as a control since these bacteria are used to feed the nematodes (Fig. 1B). The strains were selected in Dahan et al.10 based on having extreme differences in protective phenotype amongst the biological replicates in King et al.19. These phenotypic differences are well-defined and supported by genomic and biochemical analyses, as in King et al.19. Moreover, multiple replicates of each strain treatment were conducted in Dahan et al.10. In Dahan et al.10, C. elegans was colonized with the three different strains of E. faecalis or P. mendocina. Then, the worms were exposed to a complex compost microbiota and subsequent 16S rRNA sequencing was carried out (Fig. 1B). Datasets were generated by amplification of the 16S rRNA hypervariable region four (V4) using the barcoded universal primers 515F/806R and sequenced in an Illumina MiSeq system10. The raw sequence data are available in the EMBL-EBI repository under the project accession number PRJEB26987.
Analysis of 16S rRNA amplicon sequences
For this study, the 16S rRNA sequences were downloaded from EMBL-EBI repository. Using DADA2 software27 implemented in QIIME2 version 2019.728, 16S rRNA sequences were first demultiplexed and then quality trimmed based on the average quality per base of the forward and reverse reads. The first 23 nucleotides were truncated, and the total length was trimmed at 220 and 205 in forward and reverse reads, respectively. Consequently, reads were merged and chimeric variants were removed. The resulting representative sequences were taxonomically assigned using a pre-trained naïve Bayes taxonomic classifier29 based on SILVA database version 13230 and the 515F/806R primer set. The resulting taxonomic data tables were collapsed at genus level and taxa with less than 10 total reads and presents in less than 30% of samples of each dataset were removed. The taxonomic data tables were used for network analysis, keystone taxa identification and metagenome prediction (Fig. 1C).
Construction of bacterial co-occurrence networks, identification of keystone taxa and attack tolerance test
Co-occurrence network analyses were performed using the Sparse Correlations for Compositional data (SparCC) method31 implemented in R studio32. Taxonomic data tables were used to calculate the correlation matrix. Correlation coefficients with magnitude > 0.5 or < − 0.5 were selected. Network visualization and calculation of topological features and taxa connectedness (i.e., number of nodes and edges, modularity, network diameter, average degree, weighted degree, clustering coefficient and centrality metrics) was performed using the software Gephi 0.9.233. To infer keystone taxa, we used the combination of three parameters as previously described14,15: (i) eigencentrality (index that indicates the influence of the node on the co-occurrence network) values equal or higher than 0.75, (ii) clr-based abundance higher than the mean clr value (i.e., 6), and (iii) ubiquitousness, (i.e., presence of the bacteria in 100% of the samples of a given experimental condition). Scatter plots were done using the software GraphPad 8 Prism (GraphPad Software Inc., San Diego, CA, USA). The robustness of co-occurrence networks was tested with an attack tolerance test using the package NetSwan for R34. For this, networks were subjected to systematic removal of nodes using three different scenarios: (i) a random attack with 100 iterations, (ii) a directed attack where nodes are removed in decreasing order of their betweenness centrality (BNC) value (i.e., number of times a node is found on the shortest path between other nodes) and (iii) a cascading attack where BNC value are recalculated after each node removed.
Differential network analysis
Comparison of the similarity of the most central nodes between two networks was done with the package “NetCoMi”35 in R studio using the read count taxonomic tables. “Most central” nodes are defined as those nodes with a centrality value above the empirical 75% quartile. The comparison returns Jaccard's indexes for each of four local measures (i.e., degree, betweenness centrality, closeness centrality, eigenvector centrality) of the sets of most central nodes as well as for the sets of hub taxa between the two networks compared. Thus, the Jaccard’s index express the similarity of the sets of most central nodes as well as the sets of hub taxa between the two networks. Jaccard index of 0 indicates completely different sets while a value of 1 indicates equal sets of most central nodes or hub taxa between the compared networks35.
Prediction of functional traits in C. elegans microbiome
PICRUSt224 was used to predict the functional profile of bacterial communities based on 16S rRNA gene sequences. Briefly, amplicon sequence variants (ASVs) were placed into a reference tree (NSTI cut-off value of 2), which was then used to infer gene family copy numbers for each ASV. The Kyoto Encyclopedia of Genes and Genomes (KEGG) orthologs (KO)36, Enzyme Classification numbers (EC) and Cluster of Orthologous Genes (COGs)37 were used for the functional annotations of enzymes and pathways. With enzyme information, pathway profiles were inferred using structured pathway mapping based on the MetaCyc database38. Metacyc pathway was calculated based on the predicted EC number abundances.
Statistical analysis
Taxonomic, pathway and enzyme data tables, which consisted of sequencing-read counts or inferred gene families and functional gene abundance respectively were used as input of the R package ‘ALDEx2’39 for differential abundance comparisons. To avoid bias in the differential analyses due to compositional nature of the sequence data, ALDEx2 method performs centered log-ratio (clr) transformation of feature counts in each sample40. A comparison of bacterial genera, pathways or enzyme clr values was done among all the experimental groups, using Kruskal–Wallis test for multiple comparisons. For further analyses, we performed pairwise comparison between groups (i.e., protective E. faecalis vs. non-protective E. faecalis groups), which was performed using Welch's t-test. Heatmaps were built from euclidean distances of clr values to represent pathway and enzyme abundance using the R package ‘Heatplus’41.
The number of shared keystone taxa in all experimental groups, shared direct neighbor of the reference taxon Enterococcus in the three E. faecalis-exposed groups and shared differential pathways of pairwise comparisons between the three different strains of E. faecalis or P. mendocina compared to the control group was visualized using Venn diagrams implemented in the online tool http://bioinformatics.psb.ugent.be/webtools/Venn/.
Alpha and beta-diversity of functional genes were carried out on rarified EC (Enzyme commission) tables. The alpha-diversity was explored using Shannon’s diversity index42, Pielou’s evenness43 and richness44 metrics. Differences in alpha-diversity metrics between groups were tested using a Kruskal–Wallis method. Subsequently, Kruskal–Wallis pairwise comparisons between groups hypothesized to be different (i.e., protective vs. non-protective E. faecalis) was performed which served as a post-hoc test in this contest. Beta-diversity was explored using the Bray–Curtis dissimilarity and compared among all the groups using a PERMANOVA test. Betadisper function in R was used for the construction of PCoA plot based on Bray–Curtis distance matrix and an analysis of variance (ANOVA) test was used for comparison of the dispersion of the samples between the groups. For differential pathway and enzyme abundance comparison, a Kruskal–Wallis one-way ANOVA was used. In addition, a Welch’s t-test was used to determine significant pathway abundance differences in pairwise comparisons. For testing similarity of most central nodes, two p-values P(J ≤ j) and P(J ≥ j) for each Jaccard’s index, which represent the probability that the observed value of Jaccard’s index is “less than or equal” or “higher than or equal”, respectively, to the Jaccard value expected at random, were calculated. Differences were considered significant when p-value < 0.05.
Results
Impact of early colonization with symbionts on the structure of C. elegans microbiota
Ecological interactions of early bacterial colonizers with C. elegans microbiota were inferred by co-occurrence networks. Visual inspection of the networks showed distinct patterns of bacterial taxa co-occurrence in the different experimental groups (Fig. 2). The topological features of ancestral E. faecalis, non-protective E. faecalis, protective E. faecalis and P. mendocina networks reveals an increased number of edges and nodes compared with the control network, E. coli OP50 (Table 1). Jaccard index was used to test for similarities of the most central nodes in the networks. Except for the Jaccard index of betweenness centrality, the observed Jaccard index for all the other local measures were significantly higher than expected by random in the ancestral E. faecalis, non-protective E. faecalis, protective E. faecalis and P. mendocina networks compared to the control group (Supplementary Table S1). Jaccard index revealed statistical differences in several network parameters between the symbiotic and control groups which further confirms our observations of the microbial co-occurrence networks. These results suggest that the early colonization with symbionts modifies the structure of C. elegans microbial communities compared to the control communities.

Microbial co-occurrence networks of C. elegans exposed to different bacteria. Bacterial co-occurrence networks based on 16S sequences obtained from C. elegans exposed to different populations of E. faecalis (non-protective E. faecalis, protective E. faecalis and ancestral E. faecalis), P. mendocina and E. coli OP50. Nodes represent bacterial taxa and edges stand for co-occurrence correlation (SparCC > 0.5 or < − 0.5). Node size is proportional to the eigenvector centrality and node color is based on the modularity class. Thus, nodes with the same color belong to the same cluster. Gray and green edges are connecting links with negative and positive interactions, respectively. Only nodes with at least one connecting edge are displayed.
We next focused on the comparisons between microbiomes in the two evolved symbiont treatments and the ancestral treatment to study the possible impact of evolutionary history on microbe-microbe interactions. Jaccard index comparisons showed significant differences between evolved and ancestral E. faecalis networks for all the local measures, except the betweenness centrality. Only in the protective compared to the non-protective E. faecalis network, the Jaccard index for betweenness centrality was significantly higher than expected by random (Supplementary Table S1). Additionally, the network of the protective E. faecalis had the highest number of edges, nodes and average degree, while the highest number of modules was observed in the non-protective E. faecalis network (Table 1). This result suggests that early colonization with the evolved symbionts induced strain-specific modifications to the structure of C. elegans microbiota, compared to that induced by the ancestral strain.
The robustness of the networks was tested by measuring their tolerance to random or directed taxa removal. Networks of C. elegans exposed to different symbionts and subjected to random removal of taxa presented the highest tolerance, where a loss of ~ 85% of network connectivity was obtained after random removal of 50% of the nodes (Supplementary Fig. S1). Removal of taxa according to decreasing BNC produced a higher loss of connectivity in every network (Supplementary Fig. S1). However, the lowest tolerance was observed in all the networks induced by a cascading attack (Supplementary Fig. S1). Comparison of the loss of connectivity of the networks of all experimental groups showed that, independently of the type of attack, the E. coli OP50, P. mendocina and protective E. faecalis networks presented the lowest tolerances to taxa removal, followed by the ancestral E. faecalis network (Fig. 3). The non-protective E. faecalis network presented the highest tolerance to taxa removal (Fig. 3). These results suggest that the robustness of C. elegans networks is strain-specific and that the worm microbiota exposed to protective E. faecalis is as robust as the non-altered control microbiota.

Comparison of network tolerance to taxa removal based on different types of attack. The resistance of the networks to directed (left), cascading (middle) or random (right), removal of nodes was measured in non-protective E. faecalis (green line), ancestral E. faecalis (black line), E. coli OP50, P. mendocina, and protective E. faecalis (red line) networks. Nodes removal was based on their betweenness centrality (BNC) value. Loss of connectivity values range between 0 (maximum of connectivity between nodes) and 1 (total disconnection between nodes).
Impact of symbionts on keystone taxa and the hierarchical organization of C. elegans microbiota
To define changes in the hierarchical organization of C. elegans microbiota, we sought to identify keystone taxa from microbial co-occurrence networks. Ubiquitous bacteria with high eigenvector centrality values (≥ 0.75) and relative abundance (clr > 6) in the network were considered as keystone taxa. We found three and seven keystone taxa in the networks of C. elegans exposed to E. coli OP50 and the protective E. faecalis, respectively (Fig. 4). The remaining networks (ancestral E. faecalis, non-protective E. faecalis and P. mendocina) contained four keystone taxa each (Fig. 4). A comparison of the different keystone taxa in all the experimental groups showed that the protective E. faecalis network presented 3 unique keystone taxa (i.e., Clostridium sensu stricto 12, Enterococcus and Enterobacterales); E. coli OP50 network also presented one unique keystone taxa (i.e., Glunocobacter) while the remaining networks did not harbor unique keystone taxa (Fig. 4). Interestingly, the reference taxon Enterococcus became a keystone only in the protective E. faecalis group, and not in the microbial networks of the ancestral E. faecalis or non-protective E. faecalis groups. It is noteworthy that Pseudomonas was found as a keystone taxon in all networks except in the P. mendocina group (Fig. 4).

Identification of keystone taxa from microbial networks of C. elegans exposed to different bacteria. Scatter plot of the mean relative abundance (expressed as clr) vs. the eigenvector centrality of the bacterial taxa (dots or triangles) found in the microbial co-occurrence networks of C. elegans exposed to different symbionts (different E. faecalis populations or P. mendocina) and E. coli OP50. Green dots or triangles represent ubiquitous bacteria (i.e., taxa that were found across all the samples). Cutoff value of 6 were set for the mean relative abundance and 0.75 for the eigenvector centrality. Ubiquitous bacterial taxa with mean relative abundance and eigenvector centrality equal or higher than the cutoff values were considered as keystone taxa (Green triangles). List of keystone taxa is displayed next to the scatter plot. The Venn diagram shows the number of keystone taxa that are shared by or unique in the different experimental groups.
For further analysis of the microbial community structure, we constructed sub-networks containing only the keystone taxa and their direct neighbors in all the experimental conditions. We referred to them as ‘keystone sub-networks’ (Supplementary Fig. S2). Visual inspection of the keystone sub-networks showed distinct patterns of bacterial taxa co-occurrence in the different experimental groups. Bacterial members and edges of these keystone sub-networks represented less than 20% of nodes and less than 50% of the edges of the full community in all the experimental groups. Furthermore, visual inspection of each keystone sub-network showed that all keystone taxa were connected to each other. These results suggest that symbiont colonization alter the hierarchical organization of C. elegans microbiota.
Local connectivity of symbionts in C. elegans microbiota
Reference taxa (i.e., Enterococcus, Pseudomonas and Escherichia-Shigella) were defined here as the ‘genera’ taxonomic classification of the symbionts in each experimental group. The impact of early symbiont colonization on the local connectivity of reference taxa was measured using sub-networks derived from the global networks and that represent the direct neighbors around these taxa (Fig. 5). Visual inspection showed differences in the sub-network structures, reflecting potential changes in local connectivity of symbionts. Pseudomonas and Escherichia-Shigella were directly connected to only three taxa each. Enterococcus had the higher numbers of direct neighbors in the sub-networks of non-protective (22 taxa) and protective (27 taxa) E. faecalis, compared to the ancestral E. faecalis (six taxa). The results suggest that evolved symbionts establish more microbe-microbe interactions with the bacterial microbiota of C. elegans than the ancestor.

Sub-networks of the local connectivity of Enterococcus, Pseudomonas and Escherichia-Shigella in the co-occurrence networks. The direct neighbors of Enterococcus, Pseudomonas and Escherichia-Shigella were identified in the bacterial co-occurrence networks of C. elegans exposed to different strains of E. faecalis, P. mendocina as well as the food control E. coli OP50. Positive and negative interactions between co-occurring bacteria are represented by the green and gray edges, respectively. Node size is proportional to the eigenvector centrality and node color is based on the modularity class.
Further characterization of potential microbial interactions of E. faecalis was achieved by comparing the taxonomic identity of the direct neighbors of Enterococcus in the sub-networks of the three E. faecalis groups. The results revealed that the direct neighbors were mostly unique for each symbiont strain (Fig. 6A). A detailed pairwise comparison of the co-occurrence correlation between the reference taxon Enterococcus and the common taxa between the two compared groups: ancestral E. faecalis vs protective E. faecalis (Fig. 6B) and non-protective E. faecalis and protective E. faecalis (Fig. 6C) revealed that the type of connection was kept in the two comparisons. This result suggests that Enterococcus maintains common ecological associations with specific microbial members in the different groups. Changes in global (Fig. 2) and local (Fig. 5) network structure could be accompanied by hierarchical reorganization of the bacterial community.

Number and associations patterns of shared neighbors of the taxon Enterococcus. Venn diagram showing the number of bacteria that are common or unique among the taxa directly connected to Enterococcus in the ancestral E. faecalis, non-protective E. faecalis and protective E. faecalis exposure groups (A). Direction of associations of common direct neighbor between the ancestral vs. protective E. faecalis groups (B) and non-protective E. faecalis vs. protective E. faecalis groups (C). Mitochondria stands for C. elegans mitochondria.
Impact of symbionts on the functional profiles of the C. elegans microbiome
We next examined the impact of the different symbionts on the functional profiles of C. elegans microbiome. Analysis of functional gene alpha-diversity showed that C. elegans microbiome exposed to the non-protective and protective E. faecalis presented a significantly decreased functional genes richness compared to the control or P. mendocina groups (pairwise Kruskal–Wallis, p < 0.05, Supplementary Fig. S3A). Significant differences were found in the Shannon entropy of the functional genes of the protective E. faecalis group compared to the ancestral, non-protective E. faecalis and P. mendocina groups (pairwise Kruskal–Wallis, p < 0.05, Supplementary Fig. S3B). However, differences in functional genes evenness were only found between the non-protective E. faecalis and P. mendocina groups (pairwise Kruskal–Wallis, p < 0.05, Supplementary Fig. S3C). The comparison of the functional profiles (at the functional EC genes level) using PERMANOVA test on Bray–Curtis dissimilarity index showed that the microbiome of C. elegans, when exposed to different symbionts, exhibited significant differences in predicted gene abundance among all the experimental groups (PERMANOVA, p < 0.05, data no showed). Similarly, dispersion analysis of the Bray–Curtis dissimilarity index showed significant differences between the samples of the experimental groups (ANOVA, p < 0.05, Supplementary Fig. S3D).
Further analysis of the identity of the different predicted pathways revealed that 92% (i.e., 384/416) of the pathways were shared by all the groups, only one to two pathways were found exclusively in the protective E. faecalis, P. mendocina and the control group and non-unique pathway were found in the ancestral or non-protective E. faecalis groups (Supplementary Fig. S4A). Differential abundance analysis showed that several predicted pathways (Supplementary Fig. S4B) and enzymes (Supplementary Fig. S5) changed significantly among the experimental groups (Kruskal–Wallis, p < 0.05). Pairwise comparisons of the pathways between the control (E. coli OP50) and the other groups revealed those with differential abundance (Supplementary Table S2). Among the pathways with differential abundance, the protective E. faecalis group presented the highest number of unique pathways (i.e., 70), while the other experimental groups presented less than 10 unique pathways with significant differential abundance (Fig. 7). These results suggest that C. elegans microbiome exposed to the protective E. faecalis symbiont undergoes extensive functional changes and these changes are different from the other experimental groups.

Comparison of unique and share pathways across the different experimental groups. Venn diagram showing the common and unique predicted bacterial pathways, with significant abundance changes, found in the microbiome of C. elegans exposed to the ancestral E. faecalis (A), non-protective E. faecalis (B), protective E. faecalis (C) and P. mendocina (D) compared to the control group E. coli OP50 (Welch’s t-test, p < 0.05).
To study the functional departures of the evolved symbionts from the ancestral E. faecalis, we performed pairwise comparisons of the predicted pathways. Differential analysis showed no significant differences in pathway abundance between the non-protective and ancestral E. faecalis. However, three pathways were found with significant abundance differences between the protective and the ancestral E. faecalis (Supplementary Fig. S6, Welch’s t-test, p < 0.05). The abundance of two pathways, l-glutamate and L-glutamine biosynthesis (PWY-5505) and thiazole component of thiamine diphosphate biosynthesis II (PWY-6891) increased significantly while the abundance of the superpathway of thiamine diphosphate biosynthesis II (PWY-6895) decreased in the protective E. faecalis group compared to the ancestral E. faecalis group (Supplementary Fig. S6).
We next focused on the comparison between the protective and non-protective E. faecalis groups since we were interested in potential mechanisms underpinning the protective role of E. faecalis. Differential analysis showed that the abundance of three predicted pathways (Fig. 8A) and 29 enzymes (Fig. 8B) changed significantly between the protective and non-protective E. faecalis exposure groups (Welch’s t-test, p < 0.05). The abundance of two of these pathways (i.e., ergothioneine (EGT) biosynthesis I (PWY-7255), and mycolyl-arabinogalactan-peptidoglycan complex biosynthesis (PWY-6397)) decreased while the third pathway (i.e., toluene degradation III (PWY-5181)) increased significantly in the protective E. faecalis condition compared to non-protective E. faecalis group. Accordingly, the abundance of enzymes involved in the ergothioneine biosynthesis pathway such as L-histidine Nα-methyltransferase (EC 2.1.1.44) and γ-glutamyl hercynylcysteine S-oxide synthase (EC 1.14.99.50) were significantly lower in the protective E. faecalis group compared to the non-protective E. faecalis group (Supplementary Fig. S7A). The abundance of the enzyme genes N-acetylglucosaminyl-diphospho-decaprenol L-rhamnosyltransferase (EC 2.4.1.289), galactan 5-O-arabinofuranosyltransferase (EC 2.4.2.46), decaprenyl-phosphate phosphoribosyltransferase (EC 2.4.2.45), decaprenylphospho-β-D-ribofuranose 2-dehydrogenase (EC 1.1.98.3), decaprenylphospho-β-D-erythro-pentofuranosid-2-ulose 2-reductase (EC 1.1.1.333) and arabinofuranan 3-O-arabinosyltransferase (EC 2.4.2.47) involved in mycolyl-arabinogalactan-peptidoglycan complex biosynthesis were significantly lower in the protective symbiont group compared to the non-protective group (Supplementary Fig. S7B). These results highlight the impact of symbiont exposure based on phenotype on microbiome functional profiles.

Predicted enzymes and pathways with differential abundance in C. elegans exposed to highly protective and less protective E. faecalis. Heatmap showing the pathways (A) and enzymes (B) with significant differences in relative abundance (expressed as clr) between the non-protective and protective E. faecalis conditions (Welch’s t-test, p < 0.05). PWY-7255: ergothioneine biosynthesis I, PWY-6397: mycolyl-arabinogalactan-peptidoglycan complex biosynthesis, PWY-5181: toluene degradation III (aerobic) (via p-cresol).
Discussion
The disease suppressive benefits of protective microbes to their animal hosts are known45. We hypothesized that protective microbes may drive the re-structuring of the host microbial community due to complex microbe-host or microbe-microbe interactions, despite the minimal effects to the composition and diversity of the host microbiota10. Here, using available published data10, we used a network approach to assess the impact of a protective symbiont on the structure of C. elegans microbiota. Although Dahan et al.10 previously showed these symbionts had minimal impact on within-host microbiota diversity, our deep-dive into the data showed changes in community assemblages, with alterations in microbial interactions patterns and inferred functional traits. Notably, microbial co-occurrence networks can be used to identify key members of microbial communities referred as keystone taxa11,23, and we found that the greatest number of keystone taxa was identified in highly protected hosts, including the genus Enterococcus that became a keystone taxon only in the protective E. faecalis network. The keystone taxa found in the protective E. faecalis network may contribute to the microbial community resiliency to disturbance, and specifically defence against pathogens. Specific keystone bacteria have been found associated with human gut microbiome recovery after another stressor, antibiotic exposure46. Moreover, four keystone bacteria were found to maintain the functional diversity of the Ixodes scapularis microbiome under intense heat47. Whilst the results obtained here may depend on the priority placement of the protective symbionts within the host, earlier establishment of E. faecalis is not required for host-protection against pathogens in this system5,48. That said, priority effects may still give the symbiont a competitive advantage over some newly arriving commensals.
Although some attributes of the networks differed marginally between the protective E. faecalis and the other groups, such as the diameter (a measure of its cohesiveness) or the average degree, other parameters changed noticeably. Microbial networks of highly protected hosts had higher number of nodes and edges suggesting more microbial species interactions and higher complexity in host microbiota. Zheng et al. similarly found that the networks of disease-suppressive soil and healthy root of tobacco plants presented a higher number of nodes and edges and thus, higher network complexity compared to disease-conducive soil and infected roots. Another study showed that reduced microbial network complexity due to agricultural intensification was associated with decreased abundance of keystone taxa in roots49. The high complexity network may have been crucial for the suppression of soil-borne pathogens in that study12 since complex networks with interacting bacteria can enhance metabolites and information exchange. These additional interactions can make the complex network more efficient20.
The network modularity was higher in the evolved symbiont treatments compared to that of the ancestral symbiont. Modularity, which measures the strength of division of a network into modules21, can indicate network stability against disturbances such as antibiotic stress50 or climate extremes47,51. Higher complexity and modularity in the protective network could be associated with higher network robustness. To test this hypothesis, robustness was measured here as the capacity of the network to tolerate sequential removal of nodes without significant loss of network connectivity52. Our tolerance test showed that the microbiota community difference induced by protective E. faecalis colonisation, compared to that of the control (i.e., E. coli OP50), did not alter robustness. Lower robustness and higher complexity in the protective network seem counterintuitive. However, theory predicts that highly structured complexity has the risk of rare but potentially catastrophic cascading failure events initiated by small perturbations53. For example, the co-occurrence network of I. scapularis microbiota exposed to disturbance from anti-tick antibodies was highly complex with increases in the numbers of nodes and edges, but was less robust to taxa loss compared to undisturbed microbiota54. Conversely, while disturbance of the tick microbiota by infection with Anaplasma phagocytophilum, a tick-borne bacterium that protects hosts against cold stress55, increased network complexity, the robustness did not change, compared with uninfected ticks54. While establishing the interplay between network parameters and robustness is not straightforward56, our results and others54 suggest that protective symbionts can alter global network parameters with reduced impact on network robustness.
We explored the local co-occurrence of the reference taxa within nematodes (i.e., Enterococcus, Pseudomonas and Escherichia-Shigella) across different symbiont groups. We revealed higher local connectivity and stable ecological associations of Enterococcus in microbial co-occurrence networks with experimentally-evolved symbionts. This suggests that evolved symbionts form a specific community with other bacteria in the microbiota. Microbial assemblages, phylogenetically defined groups of microbes that co-occur in space and time57, can face significant barriers to assembly58. Some of these barriers include strong and positive species interactions suggesting that even stable communities may be unable to assemble58. Ecological network theory predicts that ecological dependencies between members of the microbiota make assembly predictable, but dependencies may also create barriers to assembly, as interdependent species can fail to establish when each relies on the other to colonize58. What explains the commonalities in the local assembly of microbial communities in the presence of evolved E. faecalis strains, regardless of the phenotype and/or impact for the host (non-protective vs. protective)? We consider two factors. Firstly, core microbe-microbe interactions in evolved E. faecalis (Fig. 6C) have a reduced number of positive (i.e., 2/8), and a majority of negative (i.e., 6/8) interactions, which favor community assembly, as cooperating networks of microbes can be efficient, but often unstable58,59. In diverse communities, co-occurring species with negative interactions keep one another in check, decreasing the probability of each other extinction and allowing for a stable community58. Secondly, modelling and empirical data shows that hosts can overcome barriers to assembly via mechanisms that include providing feeding58, immunity59, and differential adhesion60 to colonizers. Thus, both microbe-microbe and host-microbe interactions are important for microbiome assembly. Whether these two mechanisms act simultaneously or individually to keep the local interactions of evolved E. faecalis remains to be explored.
Comparison of the inferred functional genes of C. elegans microbiome across symbiont treatments reveals extensive disparity in functional profile. We suggest that, compared to the non-protective E. faecalis, the protective symbiont restructures the functional profiles of the microbiome. Such comparisons revealed differential abundance of metabolic pathways exclusively associated with the protective phenotype, not in vivo symbiont evolution alone. Notably, the abundance of one of these pathways, EGT biosynthesis I, decreased in the protective symbiont microbiome. EGT is a trimethylated and sulphurated histidine derivative that exhibits antioxidant properties61. Impairment of the EGT synthesis in Aspergillus fumigatus leads to a significantly reduced resistance to oxidative stress induced by elevated hydrogen peroxide61. Given that the protective role of E. faecalis in C. elegans against virulent S. aureus was mediated by superoxide production19, we hypothesize that the decrease in the antioxidant properties of the bacterial community might further enhance the oxidative stress in the nematode gut. This stress might have the effect of creating an environment even more incompatible with pathogen infection19,62,63. However, pathway prediction has the limitation of not allowing us to account for strain-specific or species-specific effects. Bacterial metagenome sequencing and/or examination of the metabolite dynamics of C. elegans microbiota during protective E. faecalis colonization may be informative for testing our hypothesis.
Beyond the general description of microbiota composition and diversity descriptors used in Dahan et al.10, we studied correlations between taxa, community structure and functionality of the microbiome across symbiont treatments. Community level analysis can grasp microbial functioning more accurately than compositional analysis alone21,64. Ecosystems are shaped by the composition of the community, but also on the type and strength of associations among co-existing members64,65,66. Network-based approaches have proven helpful in deciphering complex microbial interaction patterns21,64,65. In addition, as the community response to disturbance often involves changes in microbial interactions without affecting composition or microbial diversity, co-occurrence networks become an essential tool to compare microbial communities under different conditions51,66. For example, co-occurrence networks reveal more complexity than community composition in the resistance and resilience of plant microbial communities to drought stress66, and using a network approach revealed differential patterns of bacterial interactions in goats infected with the parasite Cryptosporidium parvum67.
Conclusions
We found that a rapidly evolved and novel protective symbiont engages in additional interactions with other colonizing micro-organisms to become a keystone taxon. The symbiont had been experimentally evolved in the absence of natural host microbiota, yet it restructured the microbial community. Experimental evolution of protective microbes could be used for microbiome engineering approaches68,69 in which microbes are ‘evolutionarily-trained’ to induce desired protective phenotypes amidst the microbiota. Such pathogen colonization resistance traits may also modify microbiota structure and restore ecological balance. Results here suggest that the mechanism of protection may matter, as protection via direct interactions with pathogens (i.e., protective E. faecalis vs. S. aureus), rather than via host-mediated immunity (P. mendocina vs. P. aeruginosa) resulted in increased keystoneness for the protective symbiont in the C. elegans microbiota. Furthermore, the hierarchical re-organization of the microbial communities may enhance the protective phenotype of E. faecalis via functional changes of C. elegans microbiota. Application of network biology will enhance our understanding of microbiota responses to protective microbial symbionts. Knowing the extent of cascading effects on microbiomes may prove helpful in determining the side-effects and effectiveness of protective symbionts, which may guide interventions to improve human and animal health.
Data availability
All material relevant to this publication are available in the manuscript. Data used to generate the figures is available in the repository Harvard Dataverse, https://doi.org/10.7910/DVN/S3SVP3.
References
Samuel, B. S., Rowedder, H., Braendle, C., Félix, M. A. & Ruvkun, G. Caenorhabditis elegans responses to bacteria from its natural habitats. Proc. Natl. Acad. Sci. USA 113, E3941–E3949 (2016).
Oliver, K. M., Smith, A. H. & Russell, J. A. Defensive symbiosis in the real world: Advancing ecological studies of heritable, protective bacteria in aphids and beyond. Funct. Ecol. 28, 341–355 (2014).
King, K. C. Defensive symbionts. Curr. Biol. 29, R78–R80 (2019).
Foster, K. R., Schluter, J., Coyte, K. Z. & Rakoff-Nahoum, S. The evolution of the host microbiome as an ecosystem on a leash. Nature 548, 43–51 (2017).
Ford, S. A., Kao, D., Williams, D. & King, K. C. Microbe-mediated host defence drives the evolution of reduced pathogen virulence. Nat. Commun. 7, 13430 (2016).
Litvak, Y. et al. Commensal Enterobacteriaceae protect against Salmonella colonization through oxygen competition. Cell Host Microbe 25, 128–139 (2019).
Pimentel, A. C., Cesar, C. S., Martins, M. & Cogni, R. The antiviral effects of the symbiont bacteria Wolbachia in insects. Front. Immunol. 11, 626329 (2021).
Becker, M. H., Brucker, R. M., Schwantes, C. R., Harris, R. N. & Minbiole, K. P. C. The bacterially produced metabolite violacein is associated with survival of amphibians infected with a lethal fungus. Appl. Environ. Microbiol. 75, 6635–6638 (2009).
Bates, K. A., Bolton, J. S. & King, K. C. A globally ubiquitous symbiont can drive experimental host evolution. Mol. Ecol. 30, 3882–3892 (2021).
Dahan, D., Preston, G. M., Sealey, J. & King, K. C. Impacts of a novel defensive symbiosis on the nematode host microbiome. BMC Microbiol. 20, 1–10 (2020).
Banerjee, S., Schlaeppi, K. & van der Heijden, M. G. A. Keystone taxa as drivers of microbiome structure and functioning. Nat. Rev. Microbiol. 16, 567–576 (2018).
Zheng, Y. et al. Exploring biocontrol agents from microbial keystone taxa associated to suppressive soil: A new attempt for a biocontrol strategy. Front. Plant Sci. 12, 655673 (2021).
Tudela, H., Claus, S. P. & Saleh, M. Next generation microbiome research: Identification of keystone species in the metabolic regulation of host-gut microbiota interplay. Front. Cell Dev. Biol. 9, 719072 (2021).
Mateos-Hernández, L. et al. Anti-tick microbiota vaccine impacts Ixodes ricinus performance during feeding. Vaccine 8, 1–21 (2020).
Mateos-Hernández, L. et al. Anti-microbiota vaccines modulate the tick microbiome in a taxon-specific manner. Front. Immunol. 12, 704621 (2021).
Dirksen, P. et al. The native microbiome of the nematode Caenorhabditis elegans: Gateway to a new host-microbiome model. BMC Biol. 14, 38 (2016).
Berg, M. et al. Assembly of the Caenorhabditis elegans gut microbiota from diverse soil microbial environments. ISME J. 10, 1998–2009 (2016).
Zhang, F. et al. Caenorhabditis elegans as a model for microbiome research. Front. Microbiol. 8, 485 (2017).
King, K. C. et al. Rapid evolution of microbe-mediated protection against pathogens in a worm host. ISME J. 10, 1915–1924 (2016).
Faust, K. & Raes, J. Microbial interactions: From networks to models. Nat. Rev. Microbiol. 10, 538–550 (2012).
Layeghifard, M., Hwang, D. M. & Guttman, D. S. Disentangling interactions in the microbiome: A network perspective. Trends Microbiol. 25, 217–228 (2017).
Röttjers, L. & Faust, K. From hairballs to hypotheses–biological insights from microbial networks. FEMS Microbiol. Rev. 42, 761–780 (2018).
Agler, M. T. et al. Microbial hub taxa link host and abiotic factors to plant microbiome variation. PLoS Biol. 14, e1002352 (2016).
Douglas, G. M. et al. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 38, 685–688 (2020).
Hou, Y. et al. Hierarchical microbial functions prediction by graph aggregated embedding. Front. Genet. 11, 608512 (2021).
Montalvo-Katz, S., Huang, H., Appel, M. D., Berg, M. & Shapira, M. Association with soil bacteria enhances p38-dependent infection resistance in Caenorhabditis elegans. Infect. Immun. 81, 514–520 (2013).
Callahan, B. J. et al. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583 (2016).
Bolyen, E. et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 7, 852–857 (2019).
Bokulich, N. A. et al. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome 6, 1–17 (2018).
Yarza, P. et al. Uniting the classification of cultured and uncultured bacteria and archaea using 16S rRNA gene sequences. Nat. Rev. Microbiol. 12, 635–645 (2014).
Friedman, J. & Alm, E. J. Inferring correlation networks from genomic survey data. PLoS Comput. Biol. 8, e1002687 (2012).
RStudio Team. RStudio: Integrated Development for R (RStudio, PBC, 2020).
Bastian, M., Heymann, S. & Jacomy, M. Gephi: An open-source software for exploring and manipulating networks. Third International AAAI Conference on Weblogs and Social Media (2009).
Lhomme, S. NetSwan: Network Strengths and Weaknesses Analysis. R Pack Version (2015).
Peschel, S., Müller, C. L., von Mutius, E., Boulesteix, A. L. & Depner, M. NetCoMi: Network construction and comparison for microbiome data in R. Brief Bioinform. 22, bbaa290 (2021).
Kanehisa, M. Goto, S, KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 28, 27–30 (2000).
Tatusov, R. L., Galperin, M. Y., Natale, D. A. & Koonin, E. V. The COG database: A tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 28, 33–36 (2000).
Caspi, R. et al. The MetaCyc database of metabolic pathways and enzymes. Nucleic Acids Res. 46, D633–D639 (2018).
Fernandes, A. D. et al. Unifying the analysis of high-throughput sequencing datasets: Characterizing RNA-seq, 16S rRNA gene sequencing and selective growth experiments by compositional data analysis. Microbiome 2, 15 (2014).
Lin, H. & Peddada, S. D. Analysis of microbial compositions: A review of normalization and differential abundance analysis. npj Biofilms Microbiomes 6, 60 (2020).
Ploner, A. Heatplus: Heatmaps with Row and/or Column Covariates and Colored Clusters. R package version 3.2. (2021).
Shannon, C. E. A mathematical theory of communication. Bell Syst. Tech. J. 27, 379–423, 623–656 (1948).
Pielou, E. C. The measurement of diversity in different types of biological collections. J. Theor. Biol. 13, 131–144 (1966).
Fisher, R. A., Corbet, A. S. & Williams, C. B. The relation between the number of species and the number of individuals in a random sample of an animal population. J. Anim. Ecol. 12, 42 (1943).
Ford, S. A. & King, K. C. Harnessing the power of defensive microbes: Evolutionary implications in nature and disease control. PLoS Pathog. 12, e1005465 (2016).
Gibbons, S. M. Keystone taxa indispensable for microbiome recovery. Nat. Microbiol. 5, 1067–1068 (2020).
Wu-Chuang, A. et al. Thermostable keystone bacteria maintain the functional diversity of the Ixodes scapularis microbiome under heat stress. Microb. Ecol. https://doi.org/10.1007/s00248-021-01929-y (2021).
Ford, S. A. & King, K. C. In vivo microbial coevolution favors host protection and plastic downregulation of immunity. Mol. Biol. Evol. 38, 1330–1338 (2021).
Banerjee, S. et al. Agricultural intensification reduces microbial network complexity and the abundance of keystone taxa in roots. ISME J. 13, 1722–1736 (2019).
Gao, Q. et al. The microbial network property as a bio-indicator of antibiotic transmission in the environment. Sci. Total Environ. 758, 143712 (2021).
de Vries, F. T. et al. Soil bacterial networks are less stable under drought than fungal networks. Nat. Commun. 9, 3033 (2018).
de Morais, U. L. A look at the way we look at complex networks’ robustness and resilience. https://arxiv.org/ftp/arxiv/papers/1909/1909.06448.pdf (2017).
Carlson, J. M. & Doyle, J. Complexity and robustness. Proc. Natl. Acad. Sci. USA 99, 2538–2545 (2002).
Estrada-Peña, A., Cabezas-Cruz, A. & Obregón, D. Resistance of tick gut microbiome to anti-tick vaccines, pathogen infection and antimicrobial peptides. Pathogens 9, 309 (2020).
Neelakanta, G., Sultana, H., Fish, D., Anderson, J. F. & Fikrig, E. Anaplasma phagocytophilum induces Ixodes scapularis ticks to express an antifreeze glycoprotein gene that enhances their survival in the cold. J. Clin. Investig. 120, 3179–3190 (2010).
Dey, A. K., Gel, Y. R. & Poor, H. V. What network motifs tell us about resilience and reliability of complex networks. Proc. Natl. Acad. Sci. USA 116, 19368–19373 (2019).
Nemergut, D. R. et al. Patterns and processes of microbial community assembly. Microbiol. Mol. 77, 342–356 (2013).
Coyte, K. Z., Rao, C., Rakoff-Nahoum, S. & Foster, K. R. Ecological rules for the assembly of microbiome communities. PLoS Biol. 19, e3001116 (2021).
Coyte, K. Z., Schluter, J. & Foster, K. R. The ecology of the microbiome: Networks, competition, and stability. Science 350, 663–666 (2015).
McLoughlin, K., Schluter, J., Rakoff-Nahoum, S., Smith, A. L. & Foster, K. R. Host selection of microbiota via differential adhesion. Cell Host Microbe 19, 550–559 (2016).
Sheridan, K. J. et al. Ergothioneine biosynthesis and functionality in the opportunistic fungal pathogen, Aspergillus fumigatus. Sci. Rep. 6, 1–17 (2016).
Rothfork, J. M. et al. Inactivation of a bacterial virulence pheromone by phagocyte-derived oxidants: New role for the NADPH oxidase in host defense. Proc. Natl. Acad. Sci. USA 101, 13867–13872 (2004).
Gaupp, R., Ledala, N. & Somerville, G. A. Staphylococcal response to oxidative stress. Front. Cell. Infect. Microbiol. Microbiol. 2, 33 (2012).
Matchado, M. S. et al. Network analysis methods for studying microbial communities: A mini review. Comput. Struct. Biotechnol. J. 19, 2687–2698 (2021).
Jiang, D. et al. Microbiome multi-omics network analysis: Statistical considerations, limitations, and opportunities. Front. Genet. 10, 995 (2019).
Gao, C. et al. Co-occurrence networks reveal more complexity than community composition in resistance and resilience of microbial communities. Nat. Commun. 13, 3867 (2022).
Mammeri, M. et al. Cryptosporidium parvum infection depletes butyrate producer bacteria in goat kid microbiome. Front. Microbiol. 16, 548737 (2020).
Foo, J. L., Ling, H., Lee, Y. S. & Chang, M. W. Microbiome engineering: Current applications and its future. Biotechnol. J. 12, 1600099 (2017).
Inda, M. E., Broset, E., Lu, T. K. & de la Fuente-Nunez, C. Emerging frontiers in microbiome engineering. Trends Immunol. 40, 952–973 (2019).
Funding
UMR BIPAR is supported by the French Government's Investissement d'Avenir program, Laboratoire d'Excellence “Integrative Biology of Emerging Infectious Diseases” (grant no. ANR-10-LABX-62-IBEID). A.W. is supported by Programa Nacional de Becas de Postgrado en el Exterior “Don Carlos Antonio López” (grant no. 205/2018). K.C.K. is grateful for funding from the European Research Council (COEVOPRO 802242). K.A.B. acknowledges funding from St. Hilda’s College Oxford.
Author information
Authors and Affiliations
Contributions
A.C.C. and K.C.K. conceived the study. A.W.C., A.E-P and D.O. performed the analyses. A.W.C. prepared the figures. A.C.C. and D.O supervised the work. A.W.C., K.A.B., A.C.C., and K.C.K. drafted the manuscript. All authors revised and accepted the last version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wu-Chuang, A., Bates, K.A., Obregon, D. et al. Rapid evolution of a novel protective symbiont into keystone taxon in Caenorhabditis elegans microbiota. Sci Rep 12, 14045 (2022). https://doi.org/10.1038/s41598-022-18269-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-18269-7
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
