
Abstract
Zebra mussel (Dreissena polymorpha) is considered as one of the 100 most harmful IAS in the world. Traditional detection methods have limitations, and PCR based environmental DNA detection has provided interesting results for early warning. However, in the last years, the development of isothermal amplification methods has received increasing attention. Among them, loop-mediated isothermal amplification (LAMP) has several advantages, including its higher tolerance to the presence of inhibitors and the possibility of naked-eye detection, which enables and simplifies its potential use in decentralized settings. In the current study, a real-time LAMP (qLAMP) method for the detection of Dreissena polymorpha was developed and tested with samples from the Guadalquivir River basin, together with two real-time PCR (qPCR) methods using different detection chemistries, targeting a specific region of the mitochondrial gene cytochrome C oxidase subunit I. All three developed approaches were evaluated regarding specificity, sensitivity and time required for detection. Regarding sensitivity, both qPCR approaches were more sensitive than qLAMP by one order of magnitude, however the qLAMP method proved to be as specific and much faster being performed in just 9 min versus 23 and 29 min for the qPCR methods based on hydrolysis probe and intercalating dye respectively.
Similar content being viewed by others
Introduction
Dreissena polymorpha is a small freshwater bivalve mollusk commonly known as zebra mussel due to the striped pattern of its shell. It is an invasive alien species (IAS), which are non-native species deliberately or accidently introduced outside their natural habitat by human action, posing a real threat to the biodiversity, human health, or economy1. D. polymorpha is native to the Black, Caspian, and Azov seas (Ponto-Caspian region) but has already spread throughout Europe and has been quickly spreading throughout the waterways of the United States and Canada2. This species was first discovered in Spain in 2001 at the Ribarroja Reservoir (Ebro River)3,4. Since its introduction, it has invaded a great part of the Ebro River basin and widely extended to other rivers and lakes. In 2009 it was found in the Guadalquivir River basin, namely at Los Bermejales Reservoir (Granada). Then, in 2011 it invaded the Iznájar Reservoir and in 2015 it was found at La Breña (both in Córdoba province).
The rapid spreading of D. polymorpha is in mainly due to their high reproductive capacity and their wide range of tolerance to environmental conditions5. A female zebra mussel may release over 40,000 eggs in a reproductive cycle and up to one million in a spawning season. The eggs are released into the water where the fertilization takes place and, within 3–5 days, develop into free-swimming larvae called veligers, which can be transported over long distances by water currents and are the most invasive form of Dreissena6. After 2–3 weeks, the veligers begin their juvenile stage by settling down, due to the weight of their forming shells, and attaching to firm underwater surfaces using byssal threads7,8. After one year of growth, they are already able to reproduce and, as adults, they can even survive 8–10 days out of water, under cool and humid conditions7,9. This fast spreading and growing of D. polymorpha prevents the few natural predators of this species from causing a steady and long-term decline in their population levels10. When attached to hard surfaces, zebra mussels can reach densities as high as 700,000 individuals/m2, causing several economic and environmental problems4,7,11. This invasive species is capable of clogging intake pipes, grids, pumping systems and other infrastructures, as well as covering hydraulic turbines, ship motors and hulls7. They can also attach to native crustaceans, snails and other bivalves, forming dense clusters and limiting their ability to move, feed, reproduce, and eventually leading to the death of these native species5,12. In addition, each individual can filter up to one liter of water per day and, due to their high densities, it can strongly affect and change the habitat they are invading5, leaving less food for other organisms and contributing to the proliferation of aquatic plants by increasing the transparency of the water. D. polymorpha is considered by the International Union for Conservation of Nature (IUCN) as one of the 100 most harmful IAS in the world13.
There are several strategies to control zebra mussels in industrial facilities, including mechanical, chemical and biological methods14,15. Although these strategies can help eliminating the invasion temporarily, they sometimes require long shutdowns which can be very costly. In addition, these control methods cannot be directly applied to the colonized reservoirs, lakes or rivers, which remain an active point for the species. Therefore, research is gradually turning towards treating the problem in the natural aquatic environment16. However, when this species is already dispersed over a large area, its dispersion can only be slowed down by management measures to prevent further spread because, once established, it is very difficult to remove, as it is the case in Spain17. Some preventive measures currently applied to avoid the spread and dispersion of D. polymorpha include information and awareness campaigns, control and restrictions on navigation and installation of disinfection stations close to the affected water bodies3.
The implementation of effective monitoring strategies allowing the detection of this species before its establishment can prevent the spread to other water bodies. In the case of settings isolated from natural ecosystems, the early detection of zebra mussels can increase the success and reduce the costs of operations aimed to remove them. Therefore, there is an urgent need of developing methods for the early detection of invasive species18. Traditional methods relying on capture for visual inspection and identification of specimens have limitations, especially regarding the correct identification of closely related species19. Moreover, the sampling protocols typically used have low probability of detecting species unless the population density is already high20. Molecular identification of species through the analysis of environmental DNA (eDNA) has been used to detect invasive species with greater sensitivity than traditional methods21,22,23,24,25. These methods provide more accurate species distribution data and can distinguish between different invasive mussel species26, also having the potential to dramatically improve the ability for an early detection and monitoring18,19,27.
Traditionally, PCR has been used for the detection of eDNA. However, novel isothermal amplification techniques have emerged in recent years with the goal of providing an analytical solution to some of the drawbacks associated with PCR/qPCR, especially due to their simplicity and reduced thermal budget28, which are interesting features for the development of methods for a potential in situ analysis. Among them, loop-mediated isothermal amplification (LAMP) has become the most popular technique29,30,31. Its main advantages compared to PCR include being performed at constant temperature, higher specificity and the possibility for achieving naked-eye detection through turbidity observation32,33. The detection can also be performed using intercalating dyes for fluorescence measurement in real-time assays34,35.
In this work, a loop-mediated isothermal amplification (LAMP) method was developed for the detection of D. polymorpha in environmental water samples. As target, the cytochrome c oxidase sub 1 (COI) was selected. The COI is a mitochondrial gene which increases the possibility of an early detection of the presence of the IAS in the environment due to the fact that mtDNA is present in thousands of copies per cell36, at the same time, the COI marker has been identified as the marker of choice for species discrimination37,38 . There has been a worldwide effort through the bar-code of life project (www.barcodinglife.org), to characterize all living animals providing unevaluable information for species identification and therefore a large database.
Despite of its advantages, LAMP has not been explored for detection of D. polymorpha in environmental samples and, considering the higher complexity of this amplification reaction, it is very important to carefully evaluate the method in order to ensure its fitness-for-purpose, particularly in terms of sensitivity and specificity, in comparison with other techniques such as real-time PCR (qPCR) before implementing this technique as part of an strategy for early warning on decentralized settings. Therefore, the current study aims to evaluate the performance of a real-time fluorescence LAMP-based method (qLAMP) developed for the detection of D. polymorpha in water samples from the Guadalquivir River basin. In addition, the results obtained were compared with two qPCR approaches, using different detection chemistries (an intercalating dye and a hydrolysis probe), also developed for this study.
Results
Optimization of qLAMP
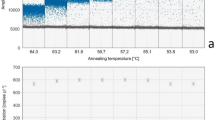
The temperature of the qLAMP assay was evaluated from 60 to 65 °C and the optimal amplification temperature resulted to be 64 °C as seen in figure S1 with a Tt of 15.9 ± 0.2 min for a sample of pure D. polymorpha DNA with a concentration of 567.3 ng µL-1. Regarding the addition of Betaine and DMSO, the best amplification results were obtained for the reactions without any supplements added, as described in Fig. 1a. The addition of DMSO promoted a significant increase in Tt, which was concentration-dependent, being 27.9 ± 0.1 min for 5% DMSO and 42.3 ± 0.1 min for 7.5% DMSO. On the other hand, the supplementation with Betaine resulted in a Tt of approximately 20 min for all concentrations tested. From this experiment, it was also observed that supplementation with DMSO had an effect on the melting temperature, resulting in a shift of the melting temperature peak from 80.9 °C down to 76.6 °C depending on the concentration, as illustrated in Fig. 1b. The concentration of LB primer was also optimized, providing a faster qLAMP reaction with a Tt of 8.8 ± 0.2 min for the same sample of pure D. polymorpha DNA, without compromising the specificity of the reaction. The Tt values were obtained using the mathematical model previously described, being represented in Table S1and Table S2.

Effect of supplement addition on the qLAMP method. (a) qLAMP kinetics in pure D. polymorpha DNA (C+) according to the fittings from Eq. (1) performed using GraphPad Prism version 8.0.0 for Windows, GraphPad Software, San Diego, California USA, www.graphpad.com: (deep red line) “C+ No supplement”, (blue line) “C+ Betaine 0.6 M”, (orange line) “C+ Betaine 0.8 M”, (purple line) “C+ Betaine 1 M”, (green line) “C+ DMSO 5%”, (yellow line) “C+ DMSO 7.5%”; (b) Respective melt curves.
Evaluation of primer specificity for qPCR and qLAMP
The specificity of the newly designed qPCR and qLAMP primers was firstly confirmed using BLAST and, later on, evaluated by testing a total of 63 environmental samples. The negative samples analyzed in this study included 17 well characterized species commonly found in the Guadalquivir River basin (12 different fish species, 1 crustacean, and 4 mollusks), up to four different individuals of each species, and five non-invaded water samples, making a total of 50 negative samples. For both qPCR and qLAMP methods, amplification was only observed for samples from D. polymorpha meat and for water samples contaminated with this species, proving the specificity of the designed primers. The positive samples analyzed included five samples from D. polymorpha meat and eight water samples from the Guadalquivir River. Two of these water samples corresponded to surface water from Los Bermejales reservoir, collected in December 2016, and from Iznájar reservoir, collected in May 2017. The other six water samples were river water in which the zebra mussels were transported, being tested as mixed water, after homogenizing the sample, and as settled water, after letting the particles settle down. Regarding the positive samples analyzed, the water sample from Los Bermejales reservoir was the only one in which no amplification neither by the qPCR approaches or the qLAMP method was observed. All the other positives samples showed amplification as expected, allowing the detection of D. polymorpha. A detailed list of the different species analyzed for specificity evaluation and the respective amplification results obtained for each method is described in Table 1.
Evaluation of sensitivity for qPCR and qLAMP
The sensitivity of the amplification methods was evaluated through the analysis of ten-fold serial dilutions of pure D. polymorpha DNA. For both qPCR approaches, positive detection was obtained from 567.3 ng µL−1 (Dil.0) down to 0.056 pg µL−1 (Dil.7), covering a dynamic range of seven orders of magnitude. Regarding the qLAMP method, positive detection was obtained from 567.3 ng µL−1 (Dil.0) down to 0.56 pg µL−1 (Dil.6), covering a dynamic range of six orders of magnitude. This corresponds to 3.992 × 105 copies µL−1 for both qPCR methods and 2.010 × 106 copies µL−1 for the qLAMP method39. All samples were tested in duplicate and the average amplification results obtained for the sensitivity evaluation of these methods are illustrated in Figs. 2 and 3, respectively.

Sensitivity evaluation through the analysis of ten-fold serial dilutions (Dil.1 – Dil.10) of pure D. polymorpha DNA with initial concentration of 567.30 ng/µL performed using GraphPad Prism version 8.0.0 for Windows, GraphPad Software, San Diego, California USA, www.graphpad.com: (red line) Dil.1, (light blue line) Dil.2, (deep red) Dil.3, (orange) Dil.4, (fucsia line) Dil.5, (purple line) Dil.6, (dark blue line) Dil.7, (dark green line) Dil.8, (green line) Dil.9, (yellow line) Dil.10. (a) qPCR method with hydrolysis probe; (b) qPCR method with F3/B3 primers. ΔRn is the magnitude of the signal generated, being given by the normalized reporter (Rn) subtracted by the baseline.

Sensitivity evaluation of the qLAMP method through the analysis of ten-fold serial dilutions (Dil.1 – Dil.10) of pure D. polymorpha DNA with initial concentration of 567.30 ng/µL according to the fittings from Eq. (1) performed using GraphPad Prism version 8.0.0 for Windows, GraphPad Software, San Diego, California USA, www.graphpad.com: (dark blue line) Dil.0, (red line) Dil.1, (light blue line) Dil.2, (deep red line) Dil.3, (orange line) Dil.4, (fucsia line) Dil.5, (purple line) Dil.6, (blue line) Dil.7, (dark green line) Dil.8, (green line) Dil.9, (yellow line) Dil.10.
Mathematical modeling
Statistical analysis of the fittings indicated the suitability of the proposed equation (Eq. 1) for modeling qLAMP kinetics (Table S1 and Table S2). In all samples, the fitted models were statistically significant, with p-values from the Fisher’s F-test lower than 0.001 and showed an excellent correlation with the experimental data (Figure S1 and Figure S2), having extremely high adjusted coefficients of determination (\(r_{adj}^{2} > 0.999\)). The estimated parameters \( RF_{{s_{max} }}\), \( \mu_{max}\) and Tt were also statistically significant (Student t-test; α = 0.05) in all amplified samples (whenever one of the replicates failed to amplify, the sample was considered negative). Tt was the parameter that exhibited more sensitivity to changes both in the type and concentration of supplements in the master mix, and to template DNA concentration. The linear range between pure D. polymorpha DNA (567.3 ng µL−1) and dilution six (0.56 pg µL−1), with Tt values ranging from 8.8 ± 0.2 min to 16.0 ± 0.2 min, is described in Fig. 4.

LOD for qLAMP in pure D. polymorpha DNA samples. Gray lines represent the 95% confidence bands for the linear estimation performed using GraphPad Prism version 8.0.0 for Windows, GraphPad Software, San Diego, California USA, www.graphpad.com.
Discussion
Due to the fast spreading of D. polymorpha, an early detection of this highly invasive species is a critical point for a more efficient management and control, making the development of methods to detect this species at low densities a high priority. The analysis of eDNA has proven to be a more effective detection method for low population densities, especially in the case of aquatic invasive species21,22,40. DNA amplification using PCR is the most widely adopted method for DNA analysis due to its high sensitivity and highly efficient exponential amplification. Despite of its advantages, this technique also has limitations, which promoted the development of alternative isothermal amplification methods such as LAMP. When compared to PCR, LAMP-based methods have higher tolerance to the presence of inhibitors, are performed at constant temperature, not requiring expensive equipment, and allow naked-eye detection, thus simplifying its implementation in point-of-need detection devices41. Several PCR methods have been developed to detect D. polymorpha through the analysis of eDNA26,42,43. However, to our knowledge, only one method has been published regarding the detection of this species using LAMP44, more studies being required in order to better evaluate the performance of this technique for the detection of invasive species in environmental samples.
In this study, a novel qLAMP method was developed and evaluated for the detection of D. polymorpha in the Guadalquivir River basin, and compared with a qPCR method, also developed in this work, using two different detection chemistries. The qLAMP method was optimized regarding primers concentration, amplification temperature and supplement addition. The optimal temperature obtained was 64 °C and the effect of supplement addition was evaluated by testing Betaine and DMSO. Betaine equalizes the contribution of G-C and A-T base pairing while DMSO disrupts base pairing, facilitating strand separation by interfering with hydrogen bonding45. Both agents are commonly used to improve LAMP sensitivity and specificity, reducing false positive results46. The reaction was supplemented with these agents in different concentrations in order to evaluate their effect. As described in Fig. 1a), the supplement addition resulted in lower LAMP efficiency, especially regarding the supplementation with DMSO, which increased the threshold time from 16 min up to 42 min. Since the addition of these agents did not improve efficiency and considering that no false positive results were obtained for any of the samples analyzed, the qLAMP method developed was performed without supplements. In addition, by optimizing the concentration of LB primer, the threshold time was reduced from 16 to 9 min, allowing a faster amplification without negatively affecting the specificity of the reaction. For the qPCR method using the hydrolysis probe for detection, the optimization was focused on primers and probe concentration, and annealing temperature. For this approach the optimal temperature resulted to be 62 °C as seen in table S3. For a better comparison between the qLAMP and qPCR methods, a qPCR using F3/ B3 primers and an intercalating dye was also evaluated, being performed with an annealing temperature of 60 °C as indicated in the manufacturer’s protocol.
The qLAMP method and both qPCR approaches were evaluated regarding primers and probe specificity. From the 63 environmental samples analyzed, amplification was only observed for positive samples, D. polymorpha meat and contaminated water samples. Therefore no false positives were obtained, proving the specificity of primers and probe, as described in Table 1. However, there was one positive water sample in which this species could not be detected neither by the qLAMP method nor the qPCR approaches. This sample corresponded to surface water collected from Los Bermejales reservoir (Guadalquivir River basin, Spain), which is known to be invaded by zebra mussels. There is a relation between the amount of eDNA and the species density, however the sampling strategy strongly influences the amount of eDNA in the samples because sampling can be done close to the species or far away from it. In addition, the degradation and dilution of eDNA is influenced by several factors, especially under natural conditions18. Another surface water sample was collected from Iznájar reservoir (Guadalquivir River basin, Spain) but, in this case, the presence of D. polymorpha was detected by all the methods tested. However, it is also important to consider that the surface water sample from Los Bermejales reservoir was collected in December, while the one from Iznájar reservoir was collected in May, which corresponds to a more active season for this species due to reproduction, thus facilitating its detection16. Zebra mussels generally reproduce when the water temperature is above 12 °C, usually during spring or summer3,16,47.
Regarding sensitivity, both detection chemistries used for qPCR were more sensitive than the qLAMP approach by one order of magnitude. In terms of speed, detection was achieved in about 9 min with qLAMP, while with the qPCR methods 23 min were needed using detection with hydrolysis probe and 29 min were needed using primers F3/ B3 and the intercalating dye. Therefore, the qLAMP method proved to be faster than both qPCR approaches, being one of the advantages of this isothermal amplification method. These DNA-based methods are both promising alternatives to the traditional microscopic analysis of the water, which is very labor intensive and time consuming.
In conclusion, a qLAMP method for the detection of D. polymorpha in environmental water samples was developed and compared with two qPCR methods using different detection chemistries. The three methods evaluated exhibited similar results in terms of specificity, allowing the detection of this species with no false positives. Regarding sensitivity, the qPCR approaches achieved better results by one order of magnitude compared to qLAMP. On the other hand, the qLAMP method proved to be much faster than both qPCR approaches, which together with its simplified thermal control, and the possibility of implementing naked-eye detection, makes this a very promising method for decentralized field analysis.
In order to improve the detection of D. polymorpha and other invasive species, further work should be done to better understand the effect of environmental and sampling conditions (e.g. season, sampling depth, water temperature) on the amount of target eDNA present in the water samples, and on the integration of detection methods with a pre-concentration step in order to move towards on-site early detection of invasive species that will surely contribute to the development of early warning systems for the presence of IAS.
Methods
Samples
Water samples from different locations including negative samples from Lima and Ave rivers as well as a fountain and a lake water sample all from Portugal, and positive samples such as transport water from D. polymorpha specimen and water from Los Bermejales and Iznajar reservoirs as well as samples from different species particularly from twelve fish species, as well as three mollusks and one crustacean species all from the Guadalquivir river provided by the company Guadalictio, except from one of the mollusks species coming from the Loire river and provided by the University of Santiago de Compostela. Additionally, five well identified D. polymorpha samples were kindly provided by Confederación Hidrogáfica del Guadalquivir.All referred samples were analyzed for the specificity evaluation of both qPCR and qLAMP approaches developed in this study. Consequently, a total of 63 samples were tested. The main goal of this evaluation was to ensure no false positive signal from other organisms, especially from those that could potentially be present in the Guadalquivir River basin. A detailed list of all the samples analyzed in this study is provided in Table 1.
DNA extraction
DNA extraction and purification from the previously mentioned samples was performed with NucleoSpin Food Kit (Macherey–Nagel, Düren, Germany) with two modifications to the protocol. These modifications were: first, the addition of 10 µL of RNase A (10 mg mL−1) in the lysis step, which was performed at 65 °C for 1 h with continuous agitation (1200 rpm), and, second, the elution was performed in two steps, adding 50 µL of Buffer CE (preheated at 70 °C) at each time. After extraction and purification, total DNA quantification was performed for each sample using the spectrophotometer NanoDrop 2000 (Thermo Scientific , Waltham, MA, United States).
Gene selection and primers design for qPCR and qLAMP
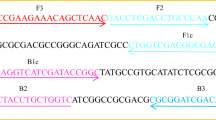
The detection of D. polymorpha was accomplished by targeting a specific region of the mitochondrial gene cytochrome C oxidase subunit I (COI). This gene has been previously used to identify D. polymorpha as well as other species, such as Dreissena bugensis, Limnoperna fortunei and Eriocheir sinensis42,48,49. A total of 40 genetic sequences for D. polymorpha COI gene were obtained from the NCBI GenBank and aligned with the CLC Sequence Viewer 7.7 software (CLC Bio-QIAGEN, Aarhus, Denmark) in order to determine a consensus sequence displayed in Fig. 5, for primer design. Primers design for the qLAMP method was performed by uploading the consensus sequence to the free software Primer Explorer V450. For the qPCR method, primers were designed with the free online software Primer351 and a hydrolysis probe was also designed for this approach. The specificity of the newly designed primers and probe was verified in silico using the Basic Local Alignment Search Tool (BLAST)52 BLAST reference number (RID) is indicated for each case in Table 2, and, later on, in vitro by analyzing the environmental samples previously described. All primers and probe were purchased from Integrated DNA Technologies Inc. (IDT, Leuven, Belgium). Detailed information regarding these oligonucleotides is described in Table 2.

Consensus sequence obtained from the alignment of 40 COI gene sequences from D. polymorpha retrieved from GenBank, and aligned with the CLC Sequence Viewer 7.7 software (CLC Bio-QIAGEN, Aarhus, Denmark).
qPCR
Two different qPCR approaches were tested in order to have a better comparison with the qLAMP method developed in this study. The first approach was performed using newly designed primers for qPCR and a hydrolysis probe, while the second one was performed using the forward and backward outer primers (F3 and B3) designed for the qLAMP method and an intercalating dye.
The primers designed for the qPCR method including a hydrolysis probe amplified a 128 bp DNA fragment with of the mitochondrial COI gene. The final reaction volume was 20 µL, with 2 µL of template DNA. In this approach, 10 µL of Maxima Probe/ROX qPCR Master Mix (Thermo Fisher Scientific Inc., Waltham, MA, USA) were used for each reaction. The reaction was optimized regarding primers and probe concentration, and amplification temperature, which was evaluated from 60 to 65 °C. The concentrations of primers and probe used were 100 nM and 150 nM, respectively. The thermal profile included an initial Uracil-DNA Glycosylase (UDG) treatment at 50 °C for 2 min and a hot start polymerase activation at 95 °C for 10 min, followed by 40 cycles of dissociation at 95 °C for 15 s and annealing-extension at 62 °C for 30 s. All experiments were carried out in a StepOne Plus Real-Time PCR system (Applied Biosystems, Foster City, CA, USA) with StepOne Software v2.1.
The second approach involved the use on an intercalating dye, in this case the F3/ B3 primers designed for qLAMP were used as primers for the qPCR, therefore targeting the same DNA fragment as the LAMP method (254 bp). The reactions were performed as detailed above but the Master Mix was replaced by Maxima SYBR Green/ ROX qPCR (Thermo Fisher Scientific Inc., Waltham, MA, USA), the primer concentration was 200 nM and the thermal profile included the UDG treatment at 50 °C for 2 min and a hot start at 95 °C for 10 min, followed by 40 cycles of dissociation at 95 °C for 15 s and annealing-extension at 60 °C for 1 min two-step cycling protocol as detailed in manufacturer’s product information. A melt curve step was included after the qPCR amplification, consisting on heating up to 95 °C for 15 s, cooling down to 60 °C for 1 min and heating up to 95 °C for 15 s, acquiring fluorescence every 0.3 °C.
Evaluation of qPCR methods
Specificity and sensitivity of both qPCR approaches were evaluated. The specificity evaluation was performed by testing the 63 environmental samples described in Table 1. The sensitivity of the methods was evaluated using ten-fold dilutions, starting from a solution of pure D. polymorpha DNA exhibiting a concentration of 567.3 ng µL−1 which corresponds to approximately 3.992 × 105 copies µL−1. All samples were tested in duplicate.
qLAMP
The newly designed LAMP primers amplified a 254 bp DNA fragment of the COI gene. All qLAMP reactions were performed with a sample volume of 2 µL to achieve a final volume of 20 µL. For each reaction, 10 µL of GspSSD Isothermal Master Mix ISO-001 (OptiGene Ltd., Horsham, UK) were used. The reaction was optimized attending to primers concentration, amplification temperature and reaction supplementation with Betaine and dimethyl sulfoxide (DMSO) (Sigma-Aldrich, Darmstadt, Germany). The amplification temperature was evaluated in the 60 °C to 65 °C range for optimization. Regarding reaction supplements, Betaine was tested at 0.6 M, 0.8 M and 1 M, while DMSO was tested at 5% and 7.5%.
The concentration of primers used was 1600 nM for FIP/BIP, 200 nM for F3/B3 and 800 nM for LB, being DNA amplification of the environmental samples performed at 64 °C for 1 h without supplements. In addition, a melt curve step was included in all qLAMP experiments, consisting of heating up to 98 °C for 15 s, cooling down to 75 °C for 1 min and heating up to 95 °C for 15 s, acquiring fluorescence every 0.3 °C.
Evaluation of qLAMP method
Sensitivity and specificity evaluation was also performed for this method in order to compare with the qPCR approaches. All samples were tested in duplicate and a sample was only considered as positive when both replicates were amplified with a threshold time (Tt) lower than 30 min.
Mathematical modeling
The qLAMP reaction was performed in a regular qPCR thermocycler (StepOne Plus Real-Time PCR). However, the equipment’s software, which is not specific for this technique, led to infra- or overestimation errors in the determination of Tt values. For this reason, the Tt values were calculated from the raw data by mathematical modeling the qLAMP kinetics, following an approach similar to that described by Garrido-Maestu et al.35. In this case, the increase of fluorescence signal (RF) as a function of time (t) was modeled as the sum of two equations:
being \(RF_{ns} \left( t \right)\) a pseudo-first order equation that accounted for the non-specific fluorescence signal (e.g. primer binding to non-target DNA):
where \(RF_{bg}\) represented the relative fluorescence background, \(RF_{{ns_{max} }}\) was the maximum non-specific fluorescence signal, and \(k_{ns}\) was the non-specific fluorescence increase rate (min−1), and being \(RF_{s} \left( t \right)\) a sigmoid equation (reparametrized Gompertz), which accounted for the target DNA amplification:
where \(RF_{{s_{max} }}\) represented the maximum specific fluorescence signal, \(\mu_{max}\) was the maximum specific amplification rate (min−1) and Tt (min) represented the threshold time at which the fluorescence intensity of the amplified DNA becomes statistically significantly higher than the background signal.
Data fitting, assessment of the model parameters significance (Student t-test; α = 0.05), and consistency of the mathematical model (Fisher’s F test; p < 0.05) were performed with Mathematica 9 (Wolfram Research, Inc.).
References
EU. No 1143/2014 of the European Parliament and of the Council of 22 October 2014 on the prevention and management of the introduction and spread of invasive alien species. Off. J. Eur. Union 2014, 35–55 (2014).
Ludyanskiy, M. L., McDonald, D. & MacNeill, D. Impact of the Zebra Mussel, a Bivalve Invader. Bioscience 43, 533–544 (1993).
Lalaguna, C. D. & Marco, A. A. The zebra mussel invasion in Spain and navigation rules. Aquat. Invasions 3, 315–324 (2008).
Rajagopal, S. et al. Origin of spanish invasion by the zebra mussel, dreissena polymorpha (pallas, 1771) revealed by amplified fragment length polymorphism (AFLP) fingerprinting. Biol. Invasions 11, 2147–2159 (2009).
CABI Invasive species Compendium: Dreissena polymorpha (zebra mussel) 2017. https://www.cabi.org/isc/datasheet/85295.
Marescaux, J. & Van Doninck, K. Using DNA barcoding to differentiate invasive Dreissena species (Mollusca, Bivalvia). Zookeys 365, 235–244 (2013).
Benson, A. J., Raikow, D., Larson, J., Fusaro, A., & Bogdanoff, A. K. Dreissena polymorpha (Pallas, 1771): U.S. Geological Survey, Nonindigenous Aquatic Species Database. Gainesville, FL (2017).
Minchin, D., Lucy, F. & Sullivan, M. Zebra Mussel: Impacts and Spread. Invasive Aquat. Species Eur. Distrib. Impacts Manag. 135–146. © 2002 Kluwer Acad. Publ. Dordreicht, Netherlands. 135–148 (2002) doi:https://doi.org/10.1007/978-94-015-9956-6_15.
Montero Melendez, J. Control of invasive alien species in Guadalquivir river basin. EURO-RIOB 2017. https://www.riob.org/es/node/4045
Molloy, D. P., Karatayev, A., Burlakova, L. E., Kurandina, D. P. & Laruelle, F. Natural enemies of zebra mussels: predators, parasites, and ecological competitors. Rev. Fish. Sci. 5, 27–97 (1997).
Nalepa, T. F. & Schloesser, D. W. Zebra Mussels Biology, Impacts, and Control (Lewis Publishers, Boca Raton, 1993).
Birnbaum, C. NOBANIS – Invasive Alien Species Fact Sheet – Dreissena polymorpha. Accessed 2 Dec 2019. https://www.nobanis.org (Online Database of the European Network on Invasive Alien Species – NOBANIS, 2011).
Lowe, S., Browne, M., Boudjelas, S., De Poorter, M. 100 of the World’s Worst Invasive Alien Species A selection from the Global Invasive Species Database. (The Invasive Species Specialist Group (ISSG), 2000).
Glomski, L. M. Zebra Mussel Chemical Control Guide. US Army Corps of Engineers: Waterways Experiment Station. https://erdclibrary.erdc.dren.mil/jspui/bitstream/11681/6966/1/ERDC-EL-TR-15-9.pdf (2015).
Boelman, S. F., Neilson, F. M., Dardeau, E. A. & Cross, T. Zebra mussel (Dreissena polymorpha) control handbook for facility operators, first edition . US Army Corps of Engineers: Waterways Experiment Station. https://hdl.handle.net/11681/2966 (1997).
Durán, C., Lanao, M., Anadón, A. & Touyá, V. Management in practice management strategies for the zebra mussel invasion in the Ebro River basin. Aquat. Invasions 5, 309–316 (2010).
Bij de Vaate, A. Rajagopal, S. & van der Velde, G. The zebra mussel in Europe: summary and synthesis. in The Zebra Mussel in Europe (ed van der Velde, G. et al.) 415–421 (Backhuys Publishers, 2010).
Herder, J. et al. Environmental DNA—a review of the possible applications for the detection of (invasive) species. Report 2013-104. Accessed 3 May 2019. https://www.researchgate.net/publication/283267157_Environmental_DNA_-_a_review_of_the_possible_applications_for_the_detection_of_invasive_species#fullTextFileContent (Stichting RAVON, Nijmegen, 2014).
Xiong, W., Li, H. & Zhan, A. Early detection of invasive species in marine ecosystems using high-throughput sequencing: technical challenges and possible solutions. Mar. Biol. 163, 1–12 (2016).
Harvey, C. T., Qureshi, S. A. & MacIsaac, H. J. Detection of a colonizing, aquatic, non-indigenous species. Divers. Distrib. 15, 429–437 (2009).
Jerde, C. L., Mahon, A. R., Chadderton, W. L. & Lodge, D. M. ‘Sight-unseen’ detection of rare aquatic species using environmental DNA. Conserv. Lett. 4, 150–157 (2011).
Dejean, T. et al. Improved detection of an alien invasive species through environmental DNA barcoding: the example of the American bullfrog Lithobates catesbeianus. J. Appl. Ecol. 49, 953–959 (2012).
Jerde, C. L. et al. Detection of Asian carp DNA as part of a Great Lakes basin-wide surveillance program. Can. J. Fish. Aquat. Sci. 70, 522–526 (2013).
Laramie, M. B., Pilliod, D. S. & Goldberg, C. S. Characterizing the distribution of an endangered salmonid using environmental DNA analysis. Biol. Conserv. 183, 29–37 (2015).
Takahara, T., Minamoto, T. & Doi, H. Using environmental DNA to estimate the distribution of an invasive fish species in ponds. PLoS ONE 8, e56584 (2013).
Gingera, T. D., Bajno, R., Docker, M. F. & Reist, J. D. Environmental DNA as a detection tool for zebra mussels Dreissena polymorpha (Pallas, 1771) at the forefront of an invasion event in Lake Winnipeg, Manitoba, Canada. Manag. Biol. Invasions 8, 287–300 (2017).
Darling, J. A. & Mahon, A. R. From molecules to management: adopting DNA-based methods for monitoring biological invasions in aquatic environments. Environ. Res. 111, 978–988 (2011).
Kaprou, G. D. et al. Miniaturized devices for isothermal DNA amplification addressing DNA diagnostics. Microsyst. Technol. 22, 1529–1534 (2016).
Mori, Y. & Notomi, T. Loop-mediated isothermal amplification (LAMP): a rapid, accurate, and cost-effective diagnostic method for infectious diseases. J. Infect. Chemother. 15, 62–69 (2009).
Fang, X., Liu, Y., Kong, J. & Jiang, X. Loop-mediated isothermal amplification integrated on microfluidic chips for point-of-care quantitative detection of pathogens. Anal. Chem. 82, 3002–3006 (2010).
Rafati, A. & Gill, P. Microfluidic method for rapid turbidimetric detection of the DNA of Mycobacterium tuberculosis using loop-mediated isothermal amplification in capillary tubes. Microchim. Acta 182, 523–530 (2014).
Notomi, T. et al. Loop-mediated isothermal amplification of DNA. Nucleic Acids Res. 28, e63 (2000).
Mori, Y., Kitao, M., Tomita, N. & Notomi, T. Real-time turbidimetry of LAMP reaction for quantifying template DNA. J. Biochem. Biophys. Methods 59, 145–157 (2004).
Tomita, N., Mori, Y., Kanda, H. & Notomi, T. Loop-mediated isothermal amplification (LAMP) of gene sequences and simple visual detection of products. Nat. Protoc. 3, 877–882 (2008).
Garrido-Maestu, A., Fuciños, P., Azinheiro, S., Carvalho, J. & Prado, M. Systematic loop-mediated isothermal amplification assays for rapid detection and characterization of Salmonella spp., Enteritidis and Typhimurium in food samples. Food Control 80, 297–306 (2017).
Verkaar, E., Nijman, I., Boutaga, K. & Lenstra, J. Differentiation of cattle species in beef by PCR-RFLP of mitochondrial and satellite DNA. Meat Sci. 60, 365–369 (2002).
Wilson-Wilde, L., Norman, J. & Robertson, J. et al. Current issues in species identification for forensic science and the validity of using the cytochrome oxidase I (COI) gene. Forensic Sci. Med. Pathol. 6, 233–241. https://doi.org/10.1007/s12024-010-9172-y (2010).
Hebert, P. D. N., Cywinska, A., Ball, S. L. & Jeremy, R. Biological identifications through DNA barcodes. Proc. Biol. Sci. 270(1512), 313–321 (2003).
Staroscik, A. Copy number calculator for realtime PCR. http://www.scienceprimer.com/copy-number-calculator-for-realtime-pcr (2012).
Ficetola, G. F., Miaud, C., Pompanon, F. & Taberlet, P. Species detection using environmental DNA from water samples. Biol. Lett. 4, 423–425 (2008).
Zanoli, L. M. & Spoto, G. Isothermal amplification methods for the detection of nucleic acids in microfluidic devices. Biosensors 3, 18–43 (2013).
Egan, S. P. et al. Rapid molecular detection of invasive species in ballast and harbor water by integrating environmental DNA and light transmission spectroscopy. Environ. Sci. Technol. 49, 4113–4121 (2015).
Xia, Z. et al. Early detection of a highly invasive bivalve based on environmental DNA (eDNA). Biol. Invasions 20, 437–447 (2018).
Williams, M. R. et al. Isothermal amplification of environmental DNA (eDNA) for direct field-based monitoring and laboratory confirmation of Dreissena sp. PLoS ONE 12, e0186462 (2017).
Frackman, B. S., Kobs, G., Simpson, D., Storts, D. & Corporation, P. Betaine and DMSO: enhancing agents for PCR. Promega Notes 65, 9–12 (1998).
Wang, D.-G., Brewster, J., Paul, M. & Tomasula, P. Two methods for increased specificity and sensitivity in loop-mediated isothermal amplification. Molecules 20, 6048–6059 (2015).
Jantz, B. & Neumann, D. Growth and reproductive cycle of the zebra mussel in the River Rhine as studied in a river bypass. Oecologia 114, 213–225 (1998).
Grigorovich, I. A., Kelly, J. R., Darling, J. A. & West, C. W. The quagga mussel invades the Lake Superior Basin. J. Great Lakes Res. 34, 342–350 (2008).
Mahon, A. R. et al. Molecular detection of invasive species in heterogeneous mixtures using a microfluidic carbon nanotube platform. PLoS ONE 6, 1–5 (2011).
PrimerExplorer. LAMP Primer Designing Software (Fujitsu Ltd, Tokyo, 2005).
Untergasser, A. et al. Primer3-new capabilities and interfaces. Nucleic Acids Res. 40, e115–e115 (2012).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990).
Acknowledgements
This work was supported by a partnership agreement project between the Confederación Hidrográfica del Guadalquivir and INL for the development of a system for early detection of zebra mussels through analysis of environmental DNA, and by project Nanotechnology Based Functional Solutions (NORTE-01-0145-FEDER-000019), supported by Norte Portugal Regional Operational Programme (NORTE2020), under the PORTUGAL 2020 Partnership Agreement, through the European Regional Development Fund. S. A. acknowledges the Portuguese funding institution FCT – Fundação para Ciência e Tecnologia for Ph.D. scholarship SFRH/BD/140396/2018. The authors would also like to thank Dr. Manuel Rey from the Department of Biochemistry and Molecular Biology from University of Santiago de Compostela (USC), the Aphanius Research Group from the department of zoology from the University of Cordoba, and, as well as to the company Guadalictio, who provided us samples of well characterized species commonly found on the Guadalquivir river basin, being used in the specificity evaluation of the detection methods developed in this study. The authors also want to thank the Water Quality Group from INL for providing non-contaminated water samples, and to Confederación Hidrográfica del Guadalquivir for providing the D. polymorpha samples used as positive controls and the contaminated river water samples.
Author information
Authors and Affiliations
Contributions
J.C. participated in all tasks related with conceptualization, methodology, validation, formal analysis, investigation, data curation, writing – original draft preparation, writing – review & edit, and visualization. A.G.M., S.A. and P.F. contributed to the conceptualization and writing – review & edit. J.B.-V. contributed to the methodology, resources, writing – review & editing, and supervision. R.J.D.M. and V.G. contributed to the conceptualization, methodology, resources and writing – review & edit. M.P. participated in the conceptualization, validation, writing – review & editing, supervision and oversaw resources, project administration and funding acquisition. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Carvalho, J., Garrido-Maestu, A., Azinheiro, S. et al. Faster monitoring of the invasive alien species (IAS) Dreissena polymorpha in river basins through isothermal amplification. Sci Rep 11, 10175 (2021). https://doi.org/10.1038/s41598-021-89574-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-89574-w
This article is cited by
-
An assessment and characterization of pharmaceuticals and personal care products (PPCPs) within the Great Lakes Basin: Mussel Watch Program (2013–2018)
Environmental Monitoring and Assessment (2024)
-
Laboratory validation of loop-mediated isothermal amplification (LAMP) assay for Limnoperna fortunei (Dunker 1857) detection
Biological Invasions (2023)
-
eDNA-based detection of the invasive crayfish Pacifastacus leniusculus in streams with a LAMP assay using dependent replicates to gain higher sensitivity
Scientific Reports (2022)
-
Dual colorimetric strategy for specific DNA detection by nicking endonuclease-assisted gold nanoparticle signal amplification
Analytical and Bioanalytical Chemistry (2022)
-
Amplified plasmonic and microfluidic setup for DNA monitoring
Microchimica Acta (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
