
Abstract
Recently, biallelic variants in the SORD gene were identified as causal for axonal hereditary neuropathy (HN). We ascertained the spectrum and frequency of SORD variants among a large cohort of Czech patients with unknown cause of HN. Exome sequencing data were analysed for SORD (58 patients). The prevalent c.757del variant was tested with fragment analysis (931 patients). Sanger sequencing in additional 70 patients was done. PCR primers were designed to amplify the SORD gene with the exclusion of the pseudogene SORD2P. Sequence differences between gene and pseudogene were identified and frequencies of SNPs were calculated. Eighteen patients from 16 unrelated families with biallelic variants in the SORD gene were found and the c.757del was present in all patients on at least one allele. Three novel, probably pathogenic, variants were detected, always in a heterozygous state in combination with the c.757del on the second allele. Patients presented with a slowly progressive axonal HN. Almost all patients had moderate pes cavus deformity. SORD neuropathy is frequent in Czech patients and the third most common cause of autosomal recessive HN. The c.757del is highly prevalent. Specific amplification of the SORD gene with the exclusion of the pseudogene is essential for a precise molecular diagnostics.
Similar content being viewed by others

Introduction
Hereditary neuropathies (HN) are a group of disorders where neuropathy is the sole or primary cause of the symptoms1 and it is already known that pathogenic variants in more than 100 genes may cause HN2. Nevertheless, the cause of the disease remains unknown for many patients and mainly for patients with primary axonal neuropathy3.
Therefore, the recent remarkable observation of biallelic pathogenic variants in the SORD gene and a new possibly treatable form of axonal hereditary neuropathy (aHN) is very significant. Moreover, the authors proposed that the prevalent variant NM_003104.6(SORD):c.757del p.(Ala253Glnfs*27); is the commonest individual pathogenic allele in the biallelic state in hereditary neuropathies4.
In the past, homozygosity mapping in consanguineous families was used for the discovery of disease causing genes for autosomal recessive (AR) hereditary neuropathies5,6. Today, the utility of whole exome sequencing (WES) in the molecular diagnostics of AR rare diseases has been demonstrated7. However, there are still many challenges as illustrated in the SORD story. Recessive causal variants might have a relatively high population frequency in GnomAD8 or similar population databases, and their pathogenic character is therefore not easy to deduce. Actually, the population frequency of the variant might be the reason that the causal gene for a recessive disorder is overlooked for a long time.
At our Centre for neuromuscular disorders, our long-lasting interest is the elucidation of the causes of hereditary neuropathies and a detailed clinical characterization of many of the autosomal recessive forms of inherited neuropathies. Hence, the description of a new form of aHN with autosomal recessive inheritance and high frequency piqued our interest. Our cohort of patients is unique since from 1997 patients from the whole of the Czech Republic have been tested in our DNA laboratory. For about 20 years, we were the sole diagnostic laboratory for HN in our country; and because of our unique position, we have been able to collect more than 3000 DNA samples from patients with HN (or probable HN), from the whole Czech Republic. To date, we have clarified the cause of HN at the DNA level in 2313 patients. In addition, our population is specific as we have one of the largest cohorts of patients with specific autosomal recessive subtypes of inherited peripheral neuropathies, for example, with disease causing variants in HINT1, SH3TC2, FIG4, HK1, NDRG1 and others9,10,11,12,13.
In this study, we performed extensive screening for the presence of SORD pathogenic variants in Czech patients with unclarified, probably hereditary peripheral neuropathies. We established the spectrum and frequencies of variants in the SORD gene in the Czech population and the relative contribution of the SORD gene to the diagnostics of HN in the Czech Republic.
Patients and methods
All patients signed an informed consent form and the ethical committee of the Motol University Hospital approved the study. All methods were carried out in accordance with relevant guidelines and regulations.
Criteria for patients selection
In total four different analyses were done. Firstly, in 58 unrelated patients with an unknown cause of HN and in whom we already had previous whole-exome sequencing (WES) data, we re-analyzed WES data for variants in the SORD gene. Secondly, we used screening with fragment analysis to search for the c.757del variant in a large cohort of 931 patients. Basically, all patients from our database, without clearly autosomal dominant inheritance and without a known cause of their disease were selected. From the 931 selected patients, 41% (387) have axonal neuropathy, 16% (152) demyelinating and 6% (52) intermediate type of HN. The information was not available for 18% (165) of the patients and the type of neuropathy was undecidable based on the available documentation in 19% (175) of patients (details are presented in the Supplementary file 1). These patients had an unknown cause of the disease, and most of the patients had been tested previously for CMT1A/HNPP or other more frequent causes of hereditary neuropathies depending on their phenotype or provided clinical and electrophysiological information. All SORD variants found by WES or fragment analysis were confirmed by Sanger sequencing in the patient and also in available family members. Finally, we continued with Sanger sequencing for additional, carefully selected patients - based on their phenotype; we searched for clinical signs described in4 - axonal type of HN (HMSN II or HMN) and compatible with recessive inheritance. In total, we sequenced exon 7 in 70 patients. From these, a subgroup of 32 patient was selected for sequencing of all remaining coding exons of the SORD gene. With this workflow, we searched for variants other than the c.757del, which is located in the exon 7.
The outline of the workflow of the study is summarized in Fig. 1. In total, four different analyses were done.
-
1.
In 58 unrelated patients with an unknown cause of HN, in whom we already had whole-exome sequencing (WES) data from the past, we re-analyzed WES data for variants in the SORD gene. If a variant was observed, it was confirmed by Sanger sequencing (three patients).
-
2.
In 931 unrelated patients with an unknown cause of HN (regardless of the type of neuropathy, but compatible with the recessive mode of inheritance, and previously tested for relevant causes of HN) screening for the presence of the c.757del variant by fragment analysis of a fluorescent PCR product was performed. A part of exon 7 was PCR-amplified with a fluorescently labelled primer specific for the SORD gene, then the fragments were separated by capillary electrophoresis and analyzed on an ABI 3130 Genetic Analyzer (ThermoFischer Scientific). The workflow, including primer sequences and the methodology, is illustrated in the Supplementary file 1. All seven c.757del homozygotes were subsequently confirmed by Sanger sequencing of exon 7 of SORD. In all of the 10 detected c.757del heterozygotes, further Sanger sequencing was done in order to identify the second causal variant. Moreover, similarly affected siblings (2 in two families) were included in the testing.
-
3.
In additional 32 unrelated patients, Sanger sequencing of all nine coding exons of the SORD gene was performed with the use of gene-specific primers. These patients were carefully selected from our database of patients based on their phenotype according to the published data; we searched for clinical signs described in4: axonal type of HN (HMSN II or HMN) and compatible with recessive inheritance.
-
4.
In additional 38 patients, Sanger sequencing of exon 7 of SORD, with the prevalent variant c.757del, was performed. This group of patients was also selected from our database according to the published clinical signs4.

Workflow of the study. Four different analyses were done. In total, biallelic SORD variants were identified in 18 patients from 16 unrelated families. Prevalent variant is the c.757del.
In total, WES data were re-analyzed for SORD in 58 patients and a fragment analysis was performed in 931 patients. Exon 7 of the SORD gene was sequenced in 92 patients in total (3 from WES; 17 from fragment analysis; 2 from additional family members in two families with similarly affected siblings; 32 patients selected from our database for Sanger sequencing of the entire coding region; and 38 different patients selected only for Sanger sequencing of exon 7 of the SORD gene). Then, exon 5 of the SORD gene was sequenced in 46 patients in total (12 heterozygotes for c.757del from WES and a fragment analysis of 1 affected sibling and 1 heterozygous carrier of c.757del from the 38 patients selected for exon 7 sequencing). In patients where the pathogenic variant on the second allele was found (c.458C>A p.(Ala153Asp); number of patients = 6) these patients were not tested further. However, for the remaining 41 patients, all 9 exons of the SORD gene were Sanger sequenced. This is summarized in Fig. 1.
For PCR amplification and Sanger sequencing, care was taken to amplify specifically the SORD gene and not the pseudogene SORD2P. To achieve this, primers for PCR amplification were designed in such a way that the primer sequence was located in a region with differences between the SORD and SORD2P sequences. In addition, it was important that primers were not located at the site of frequent polymorphism. As per the original publication4, the primers for exons 1–6 and 9 fulfilled these criteria, however, for exons 7 and 8 we recommend different primers are used.
For exon 7 of the SORD gene, the internal sequencing primer (5′-AAAAGAAAACATAGATGGCAAAAGA-3′) from the original publication is located at the site of frequent SNP: Chr15(GRCh38):g.45069181A>C and with a frequency of 21.94% in gnomAD (genomes), may cause an allele drop out that leads to a false result.
Therefore, we used a new internal sequencing primer with the sequence 5′-GCTCACGCAGCAAGCTGGTAAAG-3′ instead.
In addition, for a reliable discrimination between the gene and the pseudogene, a combination of three SNPs should be used. This is illustrated in Fig. 2. There are five differences in the sequence between SORD and SORD2P in the amplified region of exon 7. For reverse control of gene amplification, we recommend using a combination of all three SNPs, which are present in the SORD but not the SORD2P sequence.

The sequence differences of SORD and SORD2P. Blast search for SORD and SORD2P with alignment view pairwise with dots for identities (https://blast.ncbi.nlm.nih.gov). Several features are highlighted: red squares = differences; green rectangles = PCR primers; orange rectangles = sequencing primer; blue arrow = start and end of the exon; 1st primer = primer from original study; 2nd primer = our primer; grey rectangles with % = population frequency of that SNP in gnomAD for SORD.
To validate the five sequence differences in the SORD and SORD2P exon 7 sequence, we calculated the frequency of these SNPs in our patients by Sanger sequencing of exon 7.
Moreover, in the original publication, the PCR primers used for the amplification of exon 8 were not specific for SORD, as they also amplified the SORD2P exon 8. Therefore, we designed a different reverse primer to achieve a specific amplification with the sequence 5′-TCCTCCCATTCTTTAACTGGC-3′. This is illustrated in Fig. 2B.
The amplification and thermocycling protocols were used from the original paper. As for the chemistry, we used a CombiPP mix from TopBio (http://www.top-bio.cz/). For details, see the Supplementary methods section.
Research methods
All methods were carried out in accordance with relevant guidelines and regulations.
Ethics declaration
Research was conducted according to the principles expressed in Declaration of Helsinki and was approved by the local Ethics committee (University Hospital Motol). All participants or in case of children their legal representatives provided informed consent for the use of clinical data, collection of samples and subsequent DNA analyses. All patient’s data are presented in the manuscript in an anonymized form.
Results
Ballelic pathogenic, or likely pathogenic variants, in the SORD gene were detected in 18 patients from 16 unrelated families
Nine patients were homozygous for the c.757del variant p.(Ala253Glnfs*27); nine patients were compound heterozygous for c.757del and a different variant on the second allele of the SORD gene (6 × c.458C>A p.(Ala153Asp), 1 × c.218C>T p.(Ser73Leu), 1 × c.503G>A p.(Gly168Asp), 1 × c.553G>A p.(Gly185Arg)). There were six heterozygous carriers of the c.757del variant in whom no second variant was detected in the SORD gene despite Sanger sequencing of all nine SORD exons. This is illustrated in Fig. 1.
Firstly, WES data from 58 patients were reanalysed for variants in the SORD gene. Biallelic pathogenic variants were detected in three patients (5.2%) - one homozygote for c.757del and two compound heterozygotes for c.757del and c.458C>A.
Secondly, a DNA fragment analysis for the presence of c.757del was performed in 931 patients. The c.757del variant was detected on at least one allele in 17 patients (1.8% for the whole group = 931, this corresponds to 4.4% among patients with axonal neuropathy = 17/387)- seven were homozygous, and 10 were heterozygous. Confirmation of c.757del with Sanger sequencing of exon 7 of the SORD gene was done for all of them.
In addition, Sanger sequencing of exon 7 was done in an additional 70 selected (32 + 38) patients from our database. Additional two c.757del heterozygotes were detected (2.8%).
At this point, 9 (incl. one sibling) homozygotes and 15 heterozygotes (incl. one sibling) for c.757del were detected by the above-mentioned tests. All 15 heterozygotes for the c.757del were further tested by exon 5 Sanger sequencing. As previously described, c.458C>A was detected in a total of six patients, always in a heterozygous state and in combination with a c.757del on the second allele. In the remaining 9 heterozygotes (15 minus 6), Sanger sequencing of all remaining exons of the SORD was done and the second probable causal variant on the second allele of the SORD gene was found in three of them (1 × c.218C>T p.(Ser73Leu), 1 × c.503G>A p.(Gly168Asp), 1 × c.553G>A p.(Gly185Arg)).
In an additional 32 selected patients, all nine coding exons of SORD were sequenced and no additional variants were detected.
The spectrum and frequency of detected SORD variants
The most frequent variant (79% of pathogenic alleles) was the previously described c.757del detected in 33 alleles. The second most frequently detected (14% of pathogenic alleles) was the c.458C>A (also previously described as pathogenic) in 6 alleles. Furthermore, three novel missense variants were detected - always on one allele only (2%) in combination with the pathogenic and prevalent c.757del variant on the second allele. All 18 patients with biallelic, pathogenic or probably pathogenic variants in SORD carry the c.757del on at least one allele (either homozygous or heterozygous state - in combination with a second pathogenic or probably pathogenic variant in SORD). Therefore testing for the prevalent variant with subsequent sequencing of all nine coding exons in heterozygotes only should be able to detect all or nearly all patients with biallelic pathogenic variants in SORD.
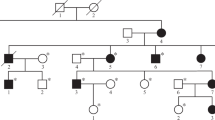
The characterization of the novel variants in exons 3, 5, and 6 is summarized in Table 1. The localization of variants in the SORD gene, electropherograms and an evaluation of conservation among the different species of these variants are presented in Fig. 3. The pedigrees of the selected families are shown in Fig. 4. In summary, nine patients were compound heterozygous for the c.757del variant and a missense variant. A segregation analysis was possible in five of them. In families 5 and 10 the parents were tested. In family 6 one offspring was tested. A segregation analysis was not possible for patients from families 7, 9, 11, 15 and 16; patients from families 7, 15 and 16 are compound heterozygous for c.[458C>A];[757del] - both variants were previously described as causal. However, a patient from family 9 is heterozygous for c.[553G>A];[757del] and a patient from family 11 is heterozygous for c.[503G>A];[757del]. A segregation analysis was not possible for these two families.
The contribution of SORD variants among other known causes of HN in Czech patients with a clarified cause of HN

Variants in the SORD gene detected in this study. Upper part - The structure of the SORD gene with nine coding exons. Middle part - Sanger sequences of variants detected in the study; detected novel variants are in pink. HET - hetrozygous; HOM-hoomzygous. Lower part - The conservation of amino acids based on a multiple sequence alignment analysis for missense variants detected in the study (https://www.ebi.ac.uk/Tools/msa/).

The pedigrees of selected families. HOM homozygous, w wild type; Pedigrees for families where at least two family members were tested are shown.
Our results, from a large collection of Czech patients with unclarified cause of presumable hereditary neuropathy, confirm that biallelic pathogenic variants in the SORD gene form a significant proportion of autosomal recessive causes of HN. Furthermore, although among Czech patients it is not the most common autosomal recessive form of HN, it is the third most frequent cause of HN (after HINT1 and the SH3TC2 variants with 27 and 24 families, respectively). The phenotype of patients with SORD bialellic variants is recognisable and we recommend screening for the prevalent c.757del variant in all unclarified HN patients, since it is an easy test to perform.
The relative contribution HN caused by biallelic variants in SORD to the whole spectrum of hereditary neuropathies with a known molecular cause among Czech patients was calculated. Since our database was established in 1997, we have clarified 2313 patients from 1171 families that have a known cause of hereditary neuropathy - with causal pathogenic variants (on average about 100 clarified patients per year). The eighteen patients we detected with biallelic SORD variants account for 0.78% (18 patients out of 2313) of all diagnosed patients and 1.36% of all families for which the cause of their neuropathy had been stated (16 families from 1171).
The clinical features of patients with the SORD neuropathy
The clinical features are summarized in the tables below.
In our patients with biallelic pathogenic variants in the SORD gene, the median age of onset was 15 years (SD ± 12.57y). The median age at examination was 55 years (SD ± 15.35y). There were 11 males and seven females; most of the patients were sporadic cases (12 families, 75%) and in four families, there were similarly affected siblings (Table 2).
Clinically, we observed slowly progressive distal peripheral neuropathy, primarily axonal, on all extremities (Table 3). All patients presented with axonal neuropathy, motor and sensory (HMSN II) or initially, and in younger patients, as pure motor axonal neuropathy (dHMN) or as an intermediate form of HMSN. Pes cavus was present in almost all adult patients (14/15). For one patient no signs of foot deformity were observed at the age of 38 years. Moreover, distal muscle weakness and atrophy were the main neuropathic symptoms, with atrophy of the calves present in all patients examined. For some patients, hypotrophy of the thighs was also prominent (4/11). Where patient information was available, the reflexes L2-L4 were decreased, and the L5-S2 were absent in most of the patients.
Discussion
The differentiation of the SORD and SORD2P sequences
The SORD gene has a pseudogene, SORD2P, with high homology (the identity is 2295 out of 2320 bases with three gaps). Pseudogenes are usually not actively transcribed or translated, and they can be recognised by the presence of a stop codon or a frameshift that interrupts the open reading frame14,15. This is true for SORD2P where the c.757del variant is normally present, and thus results in a premature stop codon and a non-functional protein. In addition, this supports the idea that the c.757del variant in the SORD gene results in a non-functional protein, as well.
However, in molecular genetic testing and diagnostics, reliable elimination of SORD2P sequences is very challenging.
In order to avoid amplification of the pseudogene, and all that entails, we adopted and validated a specific primers for PCR and Sanger sequencing. It is well known that a variation in the last three bases at the 3′ end of the primer sequence can particularly disrupt primer hybridisation and decrease PCR efficiency16. This feature can be used to obtain specific primers for genes with homologous pseudogene regions/genes. Specific primers should be chosen with mismatches at the 3′ end17. On the other hand, because the presence of frequent SNPs at the primer site may cause allelic dropout, primers need to be designed specifically to avoid frequent SNPs (population frequency > 1%) at the primer site.
Based on this, we recommend that the internal sequencing primer for exon 7 published in the original paper4 should not be used, because there is a frequent SNP at the primer site, which may cause allele drop out and lead to incorrect results. Furthermore, published primers for exon 8 of the SORD gene also co-amplify the pseudogene, SORD2P. For exon 7, this is illustrated in Fig. 2.
Another point from the original paper should be mentioned: to discriminate between SORD and SORD2P, the authors used these two SNPs in exon 7: Chr15(GRCh38):g.45068982A>T and Chr15(GRCh38):g.45069043G>A. However, in the SORD gene in the GnomAD genome database, both SNPs have a high frequency (25.7% and 1.77% respectively). Therefore, heterozygotes and even a few homozygotes may occur. And indeed, we have seen repeatedly that our samples carried those SNPs in the SORD exon 7, and our observed frequency for Chr15(GRCh38):g.45068982A>T was 17.45%. This is illustrated in Table 4.
Based on our observation, we recommend the use of different SNPs for the discrimination of gene vs pseudogene, as using these two SNPs with a high population frequency might be confusing. Our proposed workflow is as described in the method section and illustrated in Fig. 2.
Accordingly, we recommend using different sites for reliable elimination of pseudogene sequences, or a combination of all differences that are in the amplified region. In total, there are five differences and one of them is the c.757del variant. The combination of these three SNPs might be very effective for the control of specific SORD amplification: Chr15(GRCh38):g.45069043G>A, Chr15(GRCh38):g.45069087A>G, and Chr15(GRCh38):g.45068867_45068868del (illustrated in Fig. 2) – but not Chr15(GRCh38):g.45068982A>T.
NGS data analysis
For NGS data analysis, two steps should be considered for distinguishing between the SORD gene and the SORD2P pseudogene. These are: define the region of interest (target) properly (proper selection of the target region is crucial) and optimize the parameters in the analysis pipeline. However, amplification and hybridisation are done most often with commercially available kits, so it is not possible to optimize them in such a way as to improve the discrimination between genes and a pseudogene. In addition, sequencing with short reads (for ex. Illumina) is prone to ambiguous mapping, but this might be improved with pair-end sequencing which is highly recommended. The analysis pipeline has to be optimized for gene/pseudogene discrimination. In our study we applied the workflow summarized by Claes, et al.17, and three points should be highlighted here: seed length; gap penalty; and base quality score. Our NGS analysis was done according to the GATK Best practices recommendations18,19. For alignment, BWA mem was used, with seed length and gap penalty set to the default values. More information is available here: http://bio-bwa.sourceforge.net/bwa.shtml. Shorter reads are deleted and reads with gaps longer than expected are excluded. Furthermore, for base quality recalibration, we used BQSR (https://gatk.broadinstitute.org/hc/en-us/articles/360035890531-Base-Quality-Score-Recalibration) according to the GATK best practices. In every case, variants detected with NGS have to be confirmed by Sanger sequencing, and with specific PCR primers, in order to avoid coamplification of pseudogene sequences.
Our study of 18 newly detected patients with biallelic pathogenic, or probably pathogenic disease causing variants, in SORD, all with the c.757del disease causing variant on at least one allele, confirms also in a large sample of Czech patients the importance of the recent discovery of this new disease-causing gene and its prevalent disease causing variant for hereditary neuropathy4.
The recognition of the biallelic SORD variants as causal for axonal hereditary neuropathy allows for a significant increase in the diagnostic rate of aHN, especially the autosomal recessive and axonal types.
Total number of patients with SORD neuropathy and variants detected
On the other hand, as we were able to test a large number of patients from the same population and same collection of patients of a similar size, we have observed that biallelic disease causing variants in HINT1 or SH3TC2 are still a more frequent cause of HN than SORD. We have found 16 families with biallelic disease causing variants in SORD, but 27 families with HINT1 bialellic disease causing variants and 24 with SH3TC2 bialellic disease causing variants. For all three genes, we performed a very similar extensive screening of about 800 to 900 patients from our registry, collected since 1997, that had a neuropathy without a known cause. Therefore, we conclude that SORD related neuropathy is one of the most important, or the third most common, type of autosomal recessive HN, at least in the Czech population.
Clinically, the patients presented with axonal neuropathy and, in almost all of them, mild to moderate pes cavus deformity was present. Most often, the first signs of the disease occurred in their second decade of life and the disease was slowly progressive. The muscle atrophy and weakness of peroneal and calf muscles, mild to moderate foot deformities (pes cavus) are obvious. The sensory impairment of the lower limbs is relatively mild compared to the motor deficit, but the majority of patients are still able to walk unaided in the advanced stages of the disease.
To search for all possible SORD variants, and to establish the real spectrum of variants in the Czech population, we sequenced all nine coding exons of SORD in 99 Czech patients; 41 by Sanger sequencing and 58 reanalyzed using their WES data. However, no patients with biallelic pathogenic SORD variants without the c.757del in at least one copy were detected. Three novel probable pathogenic variants were detected in this study, always in combination with a prevalent c.757del variant on the second allele.
The prevalent pathogenic disease causing variant c.757del was present in all 18 patients with a biallelic pathogenic variant on at least one allele (heterozygous or homozygous state).The same was described in the original study with 45 individuals. Therefore, by screening for the c.757del only we should be able to detect all or nearly all patients with a SORD neuropathy.
In our study, four different analyses were done. WES data from 58 patients were reanalysed and three patients turned out to carry biallelic pathogenic SORD variants (5.2%). However, this is a very carefully preselected group of patients, as WES is done only after thorough full evaluation. Results from the fragment analysis with 931 patients tested represent, in our opinion, much better the true distribution of SORD variants in Czech patients with neuropathy. From a group of all unclarified patients without clear autosomal dominant inheritance, 17 carry the c.757del on one allele at least (1.8%). In addition, from these 17 patients, six were heterozygous carriers without second causal variant found (0.6%) and 11 patients have biallelic causal SORD variants (1.2%). If further larger scale studies are done, it might be interesting to see, if this is true also for other populations.
Other published cases of SORD neuropathy
Recently, biallelic SORD variants were described in four novel studies. Yuan, et al. described three patients, two homozygous for the c.757del variant and one compound heterozygous for the c.757del and c. 625C>T variant which lead to p.(Arg209*)20. Clinically, patients presented with axonal neuropathy, lower limb weakness and atrophy. The first signs of the disease occurred at puberty (14–16 years). The study by Dong et al. presented four Chinese patients21. Three of them are homozygous for the c.757del variant and one is compound heterozygous for c.404A>G p.(His134Arg) and c.908 + 1G>C, which is a splicing variant. In addition, the authors showed, that the variant results in the abolition of the donor splice site. The phenotype is very similar to other published cases with onset in the second life decade. Lower limb weakness and distal muscle atrophy are the main presentation. This paper is interesting, because it is the first published case of a patient who is compound heterozygous for c.404A>G and c.908 + 1G>C without the c.757del variant. Thirdly, a comprehensive paper by Frasqet, et al. describes a mutational spectrum in a large cohort of 163 patients with distal motor hereditary neuropathy22. Five patients with biallelic SORD variants are presented; one is homozygous for the c.757del variant, and the remaining four patients, from two unrelated families, are compound heterozygous for c.757del and c.458C>A. Two sporadic cases homozygous for c.757del are described23. Patients presented with childhood disease onset and mild phenotype. These cases further support that c.757del is the prevalent pathogenic variant. Authors concluded, that further - large scale population study - is needed to establish the proper spectrum of SORD related neuropathy in China.
The phenotype of all the patients presented in abovementioned papers in quite homogenous. The onset of the disease was in the second/third life decade. Patients present with axonal neuropathy, distal muscle weakness and atrophies. All are homozygous or compound heterozygous for the c.757del except for one patient.
All variants, detected up to date, are summarized in Fig. 5.

The SORD gene structure and published variants. Above the gene: variants found in this study. Below the gene are previously published variants. Red flags describe novel variants found in this study.
Summary
Our results show that biallelic pathogenic variants in the SORD gene are a very important cause of HN in Czech patients and are one of the most common causes of autosomal recessive HN. The c.757del disease causing variant is also highly prevalent in the Czech population and this can be used for very effective diagnostics. Our study confirms the significance of biallelic pathogenic variants in the SORD gene for hereditary neuropathies worldwide. Because of the high prevalence of the c.757del disease causing variant, we recommend screening of this variant in all patients without clear dominant family transmission, regardless the type of neuropathy. For Sanger sequencing, special care should be taken to reliably and specifically amplify only the SORD gene and elimiante the pseudogene SORD2P. For NGS data analysis, proper analysis parameters must be tweaked, especially in regard to alignment settings.
Key findings
-
1.
It is important to discriminate precisely between the SORD and SORD2P sequences. We present a suitable workflow and discuss the possible pitfalls of SNPs used for discrimination. Such a comparison has not been published or discussed, up till now.
-
2.
Biallelic variants in the SORD gene are a frequent cause of hereditary neuropathy. SORD neuropathy is one of the most important types of autosomal recessive HN. We did extensive screening and tested the largest cohort for the presence of the prevalent c.757del variant. Nobody so far has tested such a large cohort of patients with HN for the presence of the c.757del variant.
-
3.
The phenotype is quite uniform: Axonal neuropathy, with muscle weakness and distal lower limb atrophy is present in almost all patients. The onset of the disease is usually in the second or third life decade. This is concordant with other published studies.
Data availability
All protocols are available upon request. Online resources: https://blast.ncbi.nlm.nih.gov, https://www.ebi.ac.uk/Tools/msa/.
References
Reilly, M. M. Classification and diagnosis of the inherited neuropathies. Ann. Indian Acad. Neurol. 12, 80–88 (2009).
Timmerman, V., Strickland, A. V. & Zuchner, S. Genetics of Charcot-Marie-tooth (CMT) disease within the frame of the human genome project success. Genes (Basel) 5, 13–32 (2014).
Mendoza-Ferreira, N. et al. De Novo and inherited variants in GBF1 are associated with axonal neuropathy caused by golgi fragmentation. Am. J. Hum. Genet. 107, 763–777 (2020).
Cortese, A. et al. Biallelic mutations in SORD cause a common and potentially treatable hereditary neuropathy with implications for diabetes. Nat. Genet. 52, 473–481 (2020).
Azzedine, H. et al. PLEKHG5 deficiency leads to an intermediate form of autosomal-recessive Charcot-Marie-Tooth disease. Hum. Mol. Genet. 22, 4224–4232 (2013).
Senderek, J. et al. Mutations in a gene encoding a novel SH3/TPR domain protein cause autosomal recessive Charcot-Marie-Tooth type 4C neuropathy. Am. J. Hum. Genet. 73, 1106–1119 (2003).
Elmas, M. et al. Comparison of clinical parameters with whole exome sequencing analysis results of autosomal recessive patients; a center experience. Mol. Biol. Rep. 46, 287–299 (2019).
Karczewski, K. J. et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443 (2020).
Lassuthova, P., Beharka, R., Krutova, M., Neupauerova, J. & Seeman, P. COX6A1 mutation causes axonal hereditary motor and sensory neuropathy: The confirmation of the primary report. Clin. Genet. 89, 512–514 (2016).
Lassuthova, P. et al. Mutations in HINT1 are one of the most frequent causes of hereditary neuropathy among Czech patients and neuromyotonia is rather an underdiagnosed symptom. Neurogenetics 16, 43–54 (2015).
Lassuthova, P. et al. High frequency of SH3TC2 mutations in Czech HMSN I patients. Clin. Genet. 80, 334–345 (2011).
Lassuthova, P. et al. Congenital cataract, facial dysmorphism and demyelinating neuropathy (CCFDN) in 10 Czech Gypsy children–frequent and underestimated cause of disability among Czech Gypsies. Orphanet J. Rare Dis. 9, 46 (2014).
Safka Brozkova, D. et al. HMSN Lom in 12 Czech patients, with one unusual case due to uniparental isodisomy of chromosome 8. J. Hum. Genet. 62, 431–435 (2017).
Chen, X. et al. Re-recognition of pseudogenes: From molecular to clinical applications. Theranostics 10, 1479–1499 (2020).
Xu, J. & Zhang, J. Are human translated pseudogenes functional?. Mol. Biol. Evol. 33, 755–760 (2016).
Sommer, R. & Tautz, D. Minimal homology requirements for PCR primers. Nucleic Acids Res. 17, 6749 (1989).
Claes, K. B. & De Leeneer, K. Dealing with pseudogenes in molecular diagnostics in the next-generation sequencing era. Methods Mol. Biol. 1167, 303–315 (2014).
DePristo, M. A. et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 43, 491–498 (2011).
Van der Auwera, G. A. et al. From FastQ data to high confidence variant calls: The Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinform. 43, 111011–111033 (2013).
Yuan, R. Y., Ye, Z. L., Zhang, X. R., Xu, L. Q. & He, J. Evaluation of SORD mutations as a novel cause of Charcot-Marie-Tooth disease. Ann. Clin. Transl. Neurol. 8, 266–270 (2021).
Dong, H. L., Li, J. Q., Liu, G. L., Yu, H. & Wu, Z. Y. Biallelic SORD pathogenic variants cause Chinese patients with distal hereditary motor neuropathy. NPJ Genom. Med. 6, 1 (2021).
Frasquet, M. et al. Distal hereditary motor neuropathies: Mutation spectrum and genotype-phenotype correlation. Eur. J. Neurol. https://doi.org/10.1111/ene.14700 (2020).
Yonghzie, X. et al. Genetic and clinical features in 24 chinese distal hereditary motor neuropathy families. Front. Neurol. 11, 603003 (2020).
Acknowledgements
The authors wish to thank the patients and their families for their cooperation and support.
Funding
P.L. and D.S. are supported by MEYS 8F20002 under the frame of EJP RD, the European Joint Programme on Rare Diseases. In addition, this project has received funding from the European Union's Horizon 2020 research and innovation programme under the EJP RD COFUND-EJP N° 825575.
Author information
Authors and Affiliations
Contributions
Conceptualization: P.L.,L.S.; Data curation: P.L., D.S.; Formal analysis: L.S.; Funding acquisition: P.L.; Investigation: R.M., J.H.; Methodology: L.S., B.P.; Supervision: R.M., P.S.; Validation: L.S., B.P.; Writing—original draft: P.L.; Writing—review & editing: P.S.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Laššuthová, P., Mazanec, R., Staněk, D. et al. Biallelic variants in the SORD gene are one of the most common causes of hereditary neuropathy among Czech patients. Sci Rep 11, 8443 (2021). https://doi.org/10.1038/s41598-021-86857-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-86857-0
This article is cited by
-
Neurological update: hereditary neuropathies
Journal of Neurology (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
