
Abstract
Fine mapping and discovery of candidate genes underlying seed size are important for modern watermelon breeding. Here, by using a high-resolution genetic map and whole-genome genetic variation detection aided by genome survey sequencing, we fine mapped and discovered candidate genes for seed size in watermelon. QTL (quantitative trait locus) mapping identified two pleiotropic QTLs for seed size, namely, qSS4 and qSS6, using a high-density genetic map constructed by specific length amplified fragment sequencing. qSS6 explained 93.00%, 94.11% and 95.26% of the phenotypic variation in thousand-seed weight, seed length and seed width, respectively, and was defined as a major QTL. Then, high-coverage re-sequencing of two parental lines detected a total of 193,395 SNPs (single nucleotide polymorphisms) and 45,065 indels (insertions/deletions), which corresponded to a frequency of 534 SNPs/Mb and 124 indels/Mb. Based on the genetic variation in the two parental lines, newly developed PCR-based markers allowed the region of qSS6 to be narrowed to 55.5 kb. Three potential candidates were identified, including a known seed size regulator in rice, SRS3. Taken together, our results reveal successful rapid fine mapping and discovery of candidate genes for seed size in watermelon, which could be applied to many traits of interest in plants.
Similar content being viewed by others


Introduction
In most plants, the seed links the start and end of the life cycle, and is thus of particular importance. Seeds are formed by the coordinated growth of the maternal sporophytic and zygotic tissues1. Hundreds of genes have been identified that influence seed size by parent-of-origin effects, acting in the maternal and/or zygotic tissues, and have been discussed in several excellent reviews2,3,4,5,6. The functions and discovery methods of several seed-size regulators are similar in Arabidopsis and rice; therefore, converting basic research on seed size into practical applications in crops and horticultural plants, such as watermelon [Citrullus lanatus (Thunb.) Matsum. & Nakai var. lanatus], is promising.
Seed size is a heritable and important trait in watermelon, with small seeds preferred in fruit sold for consumption but large seeds preferred for planting and edible seeds. Therefore, revealing genes/QTLs (quantitative trait loci) of seed size is valuable for breeding watermelon cultivars with the desired seed size. Seed size in watermelon is highly diverse and can be classified into six representative groups as follows: giant seed, big seed, medium seed, small seed, micro seed and tomato seed7. The Mendelian inheritance of seed size in watermelon has been determined. Early research showed that the light-weight phenotype was monogenic dominant over the heavy-weight in watermelon8. Two recessive genes, s and l, determine the long and short seeds, respectively, and s is epistatic to l9. Results have shown that seed size is genetically controlled, as medium-sized seeds were dominant over both short and long seeds; thus, L-S- produces medium-sized seeds, llS- produces long seeds, and –ss produces short seeds. The monohybrid inheritance of medium over long for seed size was confirmed by later studies10,11. Moreover, the dominant genes Ti for tiny seed12 and ts gene for tomato seed were also described13,14, which seems contradictory to the previous findings.
Advances in molecular marker and sequencing technologies have made it possible to locate genetic markers linked to traits of interest. Several random amplified polymorphism DNA loci were identified to be loosely linked to SL (seed length) and SW (seed width)15. Prothro et al. identified 13 main-effect QTLs (M-QTLs) on four linkage groups for seed weight, SW and SL16. Of these, major M-QTLs were identified at the same location in both populations for all three traits in an overlapping region between 5.80 Mb and 6.41 Mb on chromosome 617. A major QTL for seed weight18 mapped in the same chromosomal region on chromosome 6 was described previously16,17, which suggested that this region is associated with seed size in watermelon from diverse genetic backgrounds. Another QTL for SL was detected by Meru and McGregor18. One QTL analysis for the medium seed and tomato seed phenotypes indicated a major QTL on chromosome 219. These current SSR (microsatellite/simple sequence repeat)/SNP (single nucleotide polymorphism) /CAPS (cleaved amplified polymorphic sequence)-based genetic maps are of high quality and accuracy but lack the marker density required to build high-resolution, integrated genetic and physical maps; thus, none of these QTLs have been fine mapped, not to mention the genes underlying them.
The advent of next-generation sequencing revolutionized genomic approaches to biology. These new sequencing tools are also valuable for high-density genetic map construction20,21, genome-wide genetic marker discovery and genotyping22, and so on. Previous research conducted in our laboratory constructed a high-density genetic map based on an F2 population derived from two watermelon cultivars with significant differences in several fruit and seed traits using low-coverage sequencing20. In the present study, using a high-resolution genetic map and whole-genome genetic variation detection aided by genome survey sequencing, we fine mapped and discovered candidate genes for seed size in watermelon. Specifically, the main objectives of this study were as follows: (1) genome-wide QTL mapping of seed size traits (including TSW (thousand-seed weight), SL, SW and ST (seed thickness)) was performed using linkage analysis; (2) genome-wide genetic variation in two parental lines was detected using high-coverage re-sequencing; (3) the major QTLs for seed size were fine mapped using newly developed PCR (polymerase chain reaction)-based markers in two populations; and (4) potential candidate genes were analysed.
Results
QTL analysis of seed size
Phenotypic variation in the parents and segregating population
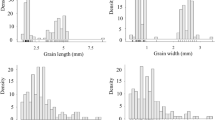
The female parent, ZXG01478, had large seeds with a TSW of 93.45 ± 9.60 g, an SL of 1.22 ± 0.03 cm, an SW of 0.70 ± 0.03 cm, and an ST of 0.20 ± 0.01 cm, whereas the male parent, 14CB11, had small seeds with a TSW of 20.80 ± 2.33 g, an SL of 0.62 ± 0.03 cm, an SW of 0.39 ± 0.01 cm, and an ST of 0.17 ± 0.01 cm (Supplementary Fig 1). Transgressive segregation was observed for all traits, indicating the presence of favourable alleles in both parents. The F1 generation gave rise to small seeds with a TSW of 30.13 ± 0.98 g, an SL of 0.68 ± 0.03 cm, an SW of 0.42 cm ± 0.01 cm, and an ST of 0.17 ± 0.01 cm, suggesting that the production of small seeds was dominant. In the F2 population, bimodal distributions for TSW, SL and SW were observed in Fig. 1, suggesting the existence of major genes for the three traits. The frequency distribution of ST in the F2 population was normal, suggesting a quantitative inheritance pattern. Correlations among TSW, SL and SW (Supplementary Table 1) were statistically significant and very strong (average r = 0.99). In contrast, the correlations between ST and the other three seed traits were also significant but somewhat lower, with an average r of 0.78 (p < 0.001).

Frequency distribution of thousand-seed weight, seed length, seed width and seed thickness in the F2 population. P1 and P2 indicate ZXG01478 (the female parent) and 14CB11 (the male parent), respectively.
QTL mapping of seed size
A high-density genetic map containing 2,634 SNP markers was previously constructed in our laboratory20. The map covered a total of 1,906.31 cM (centimorgans) of the watermelon genome and had an average distance between adjacent markers of 0.72 cM20. Genome-wide QTL scanning detected seven significant QTLs associated with TSW, SW, SL and ST (Fig. 2, Table 1). Two significant QTLs for TSW were detected on LG4 and LG6. Of these QTLs, TSW4 showed a peak LOD (logarithm of odds) score of 4.23 and explained 8.87% of the phenotypic variation, with the elite allele from the male parent, 14CB11, increasing the TSW by 11.99 g. TSW6 showed a peak LOD score of 46.92 and explained 93.00% of the phenotypic variation, with the elite allele from the female parent, ZXG01478, increasing the TSW by 42.74 g. For SL, SL4 showed a peak LOD score of 4.41 and explained 8.73% of the phenotypic variation, with the elite allele from the male parent, 14CB11, increasing the SL by 0.06 cm. Another QTL, SL6, showed a peak LOD score of 60.24 and explained 94.11% of the phenotypic variation, with the elite allele from the female parent, ZXG01478, increasing the SL by 0.30 cm. For SW, SW4 showed a peak LOD score of 4.78 and explained 10.03% of the phenotypic variation, with the elite allele from the male parent, 14CB11, increasing the SW by 0.05 cm. Another QTL, SW6, showed a peak LOD score of 48.68 and explained 95.26% of the phenotypic variation, with the elite allele from the female parent, ZXG01478, increasing the SW by 0.18 cm. Only one significant QTL for ST was identified. ST6 showed a peak LOD score of 18.05 and explained 37.92% of the phenotypic variation, with the elite allele from the female parent, ZXG01478, increasing the ST by 0.01 cm. These QTLs on LG6 for TSW, SL and SW were further confirmed by the GCIM (genome-wide composite interval mapping) method (Table 1, Fig. 2). Note that this region wasn’t associated with ST.

QTL scanning of thousand-seed weight, seed length, seed width and seed thickness. TSW, SL, SW and ST indicate thousand-seed weight, seed length, seed width and seed thickness, respectively. Black and red represents the results from CIM and GCIM, respectively.
Interestingly, three and four QTLs were co-localized to regions on LG4 and LG6, respectively, suggesting the existence of multiple closely linked QTLs or one pleiotropic QTL for these traits. To make clear this issue, two approaches were adopted. One is to use multi-trait composite interval mapping to identify QTL for the four seed size related traits. As a result, the co-localized regions on LG4 and LG6 were found to be associated with TSW, SL, SW and SL (Table 2). Meanwhile, the lowest LOD score of the co-localized region on both LG4 and LG6 was found to be associated with TSW and ST. Another is to use conditional QTL mapping. Both TSW4 and TSW6 disappeared (the LOD score was lower than the significant threshold) when SL or SW was included as a covariate in the QTL scan. Therefore, co-localized QTLs for TSW, SL and SW on LG4 and LG6 were designated as unique QTLs: qSS4 (“SS” was the abbreviation for “seed size”) and qSS6, respectively. However, when TSW was conditioned by ST, TSW6|ST was still significant (Fig. 2, Table 1), suggesting a genetic basis of TSW6 that does not entirely overlap with that of ST6.
Mining of genome-wide genetic variations
Identification and genomic distribution of SNPs and indels (insertions/deletions)
The genomic distribution of SNPs and indels was investigated based on the chromosomes of reference genome 97103. A total of 193,395 SNPs and 45,065 indels were identified between ZXG01478 and 14CB11, corresponding to a frequency of 534 SNPs/Mb and 124 indels/Mb within an approximately 362-Mb physical distance. The distributions of SNPs and indels, which were generally similar, were not evenly distributed along each chromosome (Fig. 3). The frequencies of SNPs in the 1-Mb genomic intervals were significantly positively correlated with those of indels (Spearman correlation coefficient = 0.83, p < 0.0001). The normalized average occurrence of SNPs and indels varied across the chromosomes, falling within the range of approximately 260-1,002 SNPs/Mb and 80–231 indels/Mb, respectively. The polymorphism densities were higher on chromosomes 1, 6 and 9 than on other chromosomes, and one of the two telomeres of chromosomes 6 and 9 was a particularly polymorphism-rich region. However, the polymorphism densities were lower on chromosomes 4 and 10. Based on this distribution of SNPs and indels, it was possible to construct high-density genetic maps and select SNPs and indels within specific regions for fine mapping.

Genome-wide distribution of SNPs and indels in the chromosomes of watermelon. The horizontal axis shows the chromosomes, which are divided into 1-Mb intervals. Note that 0 was assembled but not anchored to the chromosome in the reference genome 97103.
The association of predicted genes with SNPs and indels was also determined. The frequencies of genes in the 1-Mb genomic intervals were significantly positively correlated with those of SNPs (Spearman correlation coefficient = 0.39, p < 0.0001), but showed no correlation with those of indels (Spearman correlation coefficient = −0.08, p = 0.12). Moreover, the genomic distributions of genes and SSRs were investigated. The number of SSRs and genes was high at/near both ends but low in/near the middle of all the chromosomes (Supplementary Fig. 2), which likely corresponded to the peri-telomere and centromere, respectively23.
Validation of SNPs by SLAF-seq (specific length amplified fragment sequencing)
A total of 6,960 polymorphic SNPs (Supplementary Table 2) were obtained from ZXG01478 and 14CB11 by both SLAF-seq20 and the re-sequencing of two parental lines in this study. Of these SNPs, 6,712 (96.4%) were entirely consistent based on two independent sequence analyses, whereas 21 (0.3%) showed different sequences for one parental line. The remaining 227 SNPs (3.3%) showed confusing results because of uncertain sequences. A total of 2,431 SNPs anchored to the high-density genetic map were identified in the present re-sequencing analysis. Of these SNPs, 97.7% (2,375) were accurate, whereas 2.3% showed an error or uncertain sequences. Based on these data, the SNP accuracy of the present study was at least 96.4%.
Fine mapping of the major QTL, qSS6
Development of PCR-based markers in the qSS6 QTL region
The unique QTL, qSS6, showed a large effect for TSW, SL and SW and was regarded as a major QTL for seed size. To fine map this major pleiotropic QTL, the indels and SNPs in the QTL region were converted into PCR-based markers that can be rapidly and reliably analysed. To narrow down the QTL region gradually, three steps were used: first, indel markers were developed for indels with a length greater than 35 bp (base pairs); second, indel markers for indels with a length greater than 15 bp were developed based on the narrowed region; and third, CAPS and dCAPS (derived CAPS) markers were developed based on SNPs located in the re-narrowed region. Ultimately, 6, 3 and 3 indel, CAPS and dCAPS markers were developed, respectively, for further analysis (Supplementary Table 3).
Re-analysis of qSS6 after adding newly developed markers
A total of 11 newly developed PCR-based markers were genotyped in the F2 population. Of these, dcaps6_S6 and dcaps7_S6, indel14_S6 and caps4_S6, and indel16_S6 and caps5_S6 were co-segregating in the F2 population. With SNP markers on LG6, the LG6 was re-constructed using JoinMap 4.0. QTL scanning was also performed using the re-constructed LG6 and other LGs. All of the peak LOD scores for TSW, SL and SW significantly increased after the newly developed markers were added (Fig. 4, Table 1), which suggested the importance of these markers. However, the LOD score for ST showed similar results after the newly developed markers were added, which again suggested that tight linkage rather than pleiotropy was the likely genetic basis of the co-localization of ST and other seed-size traits.

The comparison of LOD scanning before (left) and after (right) adding PCR-based markers to the QTL region. The markers are in the order of the partial map of LG6.
Fine mapping of qSS6 using F2 and RIL (recombinant inbred line) populations
Using flanking markers (Marker3519 and Marker3520), we identified ten recombinants around the qSS6 locus. Using 11 newly developed co-dominant markers, a partial map of 17.22 cM was obtained (Fig. 5A). The target region was narrowed using these 10 recombinants in the F2 population (Fig. 5B). Of the six recombinants with large seeds, 13QB135–041 showed a ZXG01478 homozygous genotype to the left of indel15_S6 and was heterozygous for Marker3520, thus placing qSS6 in a region upstream of Marker3520. Similarly, 13QB135-030 placed qSS6 in a region upstream of indel15_S6, 13QB135-095 and 13QB135-007 placed qSS6 in a region downstream of Marker3519, 13QB135-005 placed qSS6 in a region downstream of caps1_S6, and 13QB135-049 delimited qSS6 to a region downstream of dcaps6_S6 and dcaps7_S6. qSS6 was narrowed to a region between dcaps6_S6 and indel15_S6 using six large-seed recombinants. Of the four recombinants with small seeds, 13QB135-028 delimited qSS6 to a region upstream of indel14_S6 and caps4_S6, 13QB135-115 placed qSS6 in a region upstream of indel16_S6 and caps5_S5, 13QB135-017 placed qSS6 in a region upstream of Marker3510, and 13QB135-020 placed qSS6 in a region downstream of indel11_S6. qSS6 was narrowed to a region between indel11_S6 and caps4_S6 using four small-seed recombinants. As a result, qSS6 was narrowed down to a 177.4-kb region between dcaps6_S6 and caps4_S6.

Graphical genotype of the selected recombinants and their seed size (including thousand-seed weight, seed length and seed width) in the F2 and RIL populations. (A) A high-resolution genetic map of the qSS6 region on LG6. The number of recombinants between adjacent markers is indicated below the map. (B) The qSS6 locus was narrowed to dcaps6_S6 and caps4_S6 by analysing the genotypes and phenotypes of the ten recombinants. (C) The qSS6 locus was delimited to a 55.5-kb interval between dcaps6_S6 and dcaps9_S6 using five flanking markers and four recombinants in the RIL population.
To further delimit qSS6, dcaps6_S6, dcaps7_S6, dcaps9_S6, indel14_S6 and caps4_S6 were used to screen recombinants in RILs, and four critical recombinants with small seeds were identified (Fig. 5C). F0032 showed a heterozygous genotype to the left of dcaps7_S6 and a ZXG01478 homozygous genotype to the right of dcaps9_S6, thus placing qSS6 in a region downstream of dcaps9_S6. F0029, F0033 and F0115 showed a 14CB11 homozygous genotype to the left of dcaps9_S6 and a ZXG01478 homozygous genotype to the right of indel14_S6, thus delimiting qSS6 to a region downstream of indel14_S6. Finally, qSS6 was narrowed to a 55.5-kb region between dcaps6_S6 and dcaps9_S6. In this interval, the LOD scores of TSW6, SL6 and SW6 were 65.62, 60.08 and 71.75, respectively, which were also the peak values. Taken together, these results indicated that qSS6 should be positioned in a 55.5-kb region between dcaps6_S6 and dcaps9_S6.
Homology and functional prediction of potential candidate genes underlying qSS6
Based on the genome sequence of watermelon (http://www.icugi.org/), four putative genes were annotated (Cla009289, Cla009290, Cla009291 and Cla009292) in the 55.5-kb interval. Protein BLAST pairwise alignment against the Arabidopsis database for Cla009289 gave significant hits with the ATL gene family, which comprises a large number of putative ubiquitin ligases of the RING-H2 type24. Cla009290 gave significant hits with AT3G06840.1, which encodes an unknown protein. Cla009291 gave significant hits with MATE (multidrug and toxic compound extrusion) efflux family protein and putatively encoded the MDR (multidrug resistance) protein mdtK. Cla009292 encoded MARD1 (Mediator of ABA-Regulated Dormancy 1), which is an important downstream component of the ABA signalling pathway that mediates ABA-regulated seed dormancy in Arabidopsis25. qRT-PCR (quantitative RT-PCR) of RILs with large and small seeds indicated that only Cla009291 was significantly differentially expressed at different seed developmental stages (Fig. 6). The marker caps5_S6, based on an SNP (G to T, in the first exon, S/end4) revealed a stop-gain effect for Cla009310. The putative gene Cla009310 contained 195 amino acids and encoded an unknown protein. Moreover, near the QTL region, one annotated gene, Cla009301, encoded BY-kinesin-like protein 10, which contains a kinesin motor domain and is a homologue of SRS3 (a gene regulating SL in rice26), was also a potential candidate gene under qSS6.

The expression profiles of potential candidate genes in seeds at different developmental stages using qRT-PCR. DAF is the abbreviation for days after flowering. Watermelon actin was used as an internal control. Data are expressed as the means of the three biological replicates; error bars indicates SDs; ** indicates significance at p < 0.001.
Discussion
Here, using a high-resolution genetic map and whole-genome genetic variation detection aided by genome survey sequencing, we fine mapped and discovered candidate genes for seed size in watermelon. By simplifying genomic data using a reduced representation library (i.e., SLAF-seq), linkage and QTL identification were performed in a cost-effective manner and revealed a major pleiotropic QTL for seed size, qSS6, in an F2 population including 93 individuals (Fig. 2). Then, new PCR-based markers were developed based on deep re-sequencing of two parental lines, and fine mapping of qSS6 was carried out to narrow the QTL to a 55.5-kb stretch flanked by dcaps6_S6 and dcaps9_S6, a segment that contained only four putative proteins, in the F2 and RIL populations (Fig. 5). One gene/QTL can be fine mapped to a small region if it exhibits the characteristics of segregation of a single Mendelian factor27 in an ideal mapping population, i.e., if the segregation pattern shows the following characteristics: (1) the frequency distribution of the trait shows clear discontinuous or bimodal segregation27,28; (2) the trait segregation exhibits a ratio of 3:1 or 1:2:129. In this study, the bimodal segregation pattern of the data for TSW, SL and SW suggested that one major QTL controlled these traits (Fig. 1) in the F2 population. Therefore, it was possible to preliminarily fine map the major QTLs in this F2 population. In experiments aimed at fine mapping genes/QTLs, it is often necessary to saturate a specific target region with a large number of markers30. A total of 193,395 SNPs and 45,065 indels were identified between two parental lines in the present study. Of the SNPs, only 3.6% (6,960) showed polymorphism as detected by SLAF-seq, which ensured a high enough marker density when fine mapping the target QTL. Therefore, we have successfully narrowed the confidence interval of the target QTL to a physical distance of 55.5 kb. Compared with the traditional NIL (near-isogenic line)-based fine mapping strategy31, this method does not require the genotyping of a large-scale NIL segregating population and is labour and time saving. This rapid fine mapping strategy highlights the direction of gene mining, which could be applied to many traits of interest in plants.
Several previous studies used different mapping populations to identify QTLs for seed size in watermelon. Prothro et al.16 identified a major QTL for seed weight on chromosome 6 in the elite × elite [flanking markers NW0251236 (5.80 Mb) and NW0250242 (6.44 Mb)] and elite × citron [flanking markers NW0248118 (5.04 Mb) and NW0248583 (6.41 Mb)] populations. Ren et al. (2014) anchored these markers to an integrated map and found that a 0.62 Mb region (from 5.80 to 6.41 Mb) overlapped between the flanking markers in these populations. Meru and McGregor 18 found that the closest marker to a major QTL for 100-seed weight, SL and SW on chromosome 6 was NW0250854 (4.58 Mb). In the present study, according to the development of indel, CAPS and dCAPS markers based on indel and SNP information for the QTL region, the pleiotropic major QTL for seed size, qSS6, was narrowed to a physical distance of 55.5 kb (from 5.37 to 5.42 Mb) on chromosome 6 and only contained only four predicted genes. Notably, the allelic variation determined by the marker dcaps9_S6 (5.42 Mb) co-segregated with seed size in the F2 population. Our results confirmed the major QTL for seed size on LG6 and further corrected its precise location compared with previous studies16,18. In addition, the overlapping of the simple confidence intervals of different QTLs might have masked the correct position17. The major QTL for tomato seed was also identified on chromosome 219. Two minor QTLs on chromosome 2 and one on chromosome 8 were identified16,17. We also identified a novel QTL for seed size on LG4.
As for the four putative candidate genes located in the narrowed qSS6 region, only Cla009291 was significantly differentially expressed at different seed developmental stages between large- and small-seeded lines. The annotated gene Cla009291 encodes the MDR protein mdtK. The MDR transporter genes are an emerging group involved in a diverse array of developmental and metabolic processes in plants. For example, the MDR-like proteins AtMDR1 and AtPGP1 are required for phytohormone auxin-mediated plant development32,33. ZmMrp3, an MDR-associated protein, is required for the anthocyanin transport process in maize34. A MATE family member in Arabidopsis thaliana, AtDTX50, functions as an ABA efflux transporter35. TT12 (TRANSPARENT TESTA12) encodes a multidrug secondary transporter-like protein required for flavonoid sequestration in vacuoles of the seed coat endothelium36. ABS3 and ABS4, a subgroup of MATE family transporters, are potential negative regulators of hypocotyl cell elongation in Arabidopsis37. The determination of exact transport substrates and how they are linked to plant development is one of the major challenges for MATE transporters. Cl009291 likely transports a small molecule(s) that is important for many aspects of seed development and may act as a seed growth regulator through its direct or indirect actions; thus, Cl009291 is an important potential candidate gene in qSS6. Some SNPs can alter a protein directly, such as non-synonymous SNPs, stopped-gained or stop-lost SNPs, frameshift SNPs or SNPs in splice sites38. A stop-gained SNP in the first exon (S/end4) of Cla009310 significantly altered the protein. Therefore, Cla009310, the gene near the narrowed qSS6 region, is also a potential candidate. In rice, one causal gene for novel small and round seed phenotypes of srs3 codes for a protein that contains a kinesin motor domain26. Comparative analysis suggested that the annotated watermelon gene Cla009301, a homologue of SRS3, encoding BY-kinesin-like protein 10, which contains a kinesin motor domain, is one of the important candidate genes for seed size in the QTL, qSS6.
In summary, using a high-resolution genetic map and whole-genome genetic variation detection aided by genome survey sequencing, we fine mapped qSS6 and identified three candidate genes. The method could be applied to many traits of interest in plants. We also found useful PCR-based markers near qSS6 that may assist in breeding for seed size and can lay the foundation for functional validation of the candidate genes to elucidate the molecular mechanism underlying seed size. In further studies, functional analysis of parental lines or NILs will be used to validate the candidate genes by sequence analysis and genetic transformation.
Methods
Plant materials, field experiments, trait evaluation and statistical analysis
A large F2 population of 536 individuals derived from a cross between ZXG01478 and 14CB11 showed significant differences in seed weight, SL and SW20. A total of 93 F2 individuals were randomly selected for genotyping and QTL analysis. A total of 122 F2 individuals were randomly selected and subjected to single-seed descent for four generations, which produced 106 RILs. Both parents, the F1s, F2 progeny and the RILs were planted at the Zhengzhou Fruits Research Institute of the Chinese Academy of Agricultural Sciences in Zhengzhou. The two parents and the F1s were grown in triplicate, with 10 plants each. Then, 93 F2 individuals and 106 RILs were planted in a green house following essentially regular breeding practices. All fruits were harvested at full maturity between 35 to 40 days after pollination, and a single fruit per plant was harvested. The seed weight of each fruit was measured based on 50 fully developed seeds. The average seed weight was then converted into TSW. The SL, SW and ST were measured using Vernier callipers based on 20 randomly selected seeds per fruit.
Pearson and Spearman correlation coefficients were calculated using SAS software39.
QTL mapping of seed size
Genome-wide QTL mapping of TSW, SL, SW and ST was performed using the composite interval mapping (CIM) program40 in WinQTL Cartographer 2.5 software (http://statgen.ncsu.edu/qtlcart/WQTLCart.htm). The LOD threshold was determined via a permutation test with 1000 repetitions41. LOD scores corresponding to p = 0.05 were used to identify significant QTLs (4.22, 4.17, 4.27 and 4.16 for TSW, SL, SW and ST, respectively). The QTL intervals were established by 2-LOD as approximately 95% QTL confidence intervals. The GCIM method42 was also conducted to validate the above detected QTLs for seed size related traits.
To distinguish multiple closely linked QTLs from one pleiotropic QTL in one narrow interval, two approaches were adopted. One is to use multi-trait composite interval mapping43 implemented in WinQTL Cartographer v2.5. Another is to use conditional QTL mapping. The conditional phenotypic values y (T1/T2) were obtained by a mixed model approach for conditional analysis of quantitative traits44 using QGAStation 1.0 (http://ibi.zju.edu.cn/software/qga/index.htm), where T1|T2 indicates that trait 1 is conditioned by trait 2. Then, the conditional mapping of the QTLs was conducted according to the conditional phenotypic values using the same method as that for the unconditional QTLs mentioned above.
Genome-wide identification of SNPs, indels and SSRs
ZXG01478 and 14CB11 were used in re-sequencing for genome-wide detection of SNPs and indels. The paired-end reads from the two parental watermelon lines were aligned to the reference genome of watermelon22 using the BWA (Burrows-Wheeler alignment tool)45. Sequence alignment file conversions were performed using SAMtools46. A total of 18.6 G of data was output from the two lines, which covered an average of 22 × of the watermelon genome. SNP and indel mining were performed using GATK (Genome Analysis Toolkit)47. The reference watermelon genome was used as a “bridge” to sequentially detect SNP and indel polymorphisms between two re-sequenced parental watermelon lines.
The identification and localization of SSRs in watermelon reference genome 97103 were accomplished by MISA (MIcroSAtellite, an SSR mining tool; http://pgrc.ipk-gatersleben.de/misa/misa.html). The minimum repeat unit was defined as 12, 6, 4, 3, 3 and 3 for mono-, di-, tri-, tetra-, penta- and hexa-nucleotides, respectively.
PCR-based marker development
Extracting 500 bp before and after each SNP/indel locus was performed with a self-compiled script in Perl. To develop PCR-based CAPS and dCAPS markers, the free Web-based software dCAP Finder 2.0 (http://helix.wustl.edu/dcaps/dcaps.html) was used to find appropriate restriction enzymes for detecting SNPs48. Primer 5.049 and Oligo 750 were used to design the respective PCR primer sets (including CAPS, dCAPS and indel markers).
Genotyping of CAPS, dCAPS and indel markers in the F2 and RIL populations
Genomic DNA was extracted from fresh leaves of the parents, F1s, F2 progeny and RILs using a modified CTAB method51. PCR was performed in 25-μl reaction volumes containing 12.5 μl 2 × Power Taq PCR MasterMix (Bioteke, Beijing, China) with 10 μM of each primer and approximately 50 ng of genomic DNA as a template. Thermocycling was started at 94 °C for 5 min, followed by 35 cycles of 94 °C for 20 s, 55 °C for 1 min and 72 °C for 30 s, with a final extension at 72 °C for 5 min. The PCR products were separated on 8% polyacrylamide gel and visualized by silver staining.
Re-linkage analysis
Re-linkage analysis was performed using JoinMap 4.0 software (http://www.kyazma.nl/index.php/mc.JoinMap) with a goodness-of-fit threshold of ≦5, a recombination frequency of <0.4 and a minimum LOD score of 2.0. All genetic distances were expressed in cM as derived by the Kosambi function52.
qRT-PCR analysis
To further narrow the candidate genes, the seeds were collected from 7 DAF (days after flowering) to 13 DAF to investigate the SL, SW and ST of the selected RILs (Supplementary Fig. 3) with large and small seeds: RIL_L (F0043) and RIL_S (F0055-1), respectively. The results showed that seeds expanded rapidly after flowering. Seeds generally expanded to their final size at 8 DAF and 12-13 DAF for RIL_L and RIL_S, respectively.
For the qRT-PCR template, the reverse transcription reaction was performed using a PrimeScriptTM II 1st Strand cDNA Synthesis Kit (Takara, Beijing, China). qRT-PCR was performed using a Bio-Rad IQ5 with SYBR Green detection. Relative expression levels were evaluated using the 2−△△CT method. The watermelon actin gene was used as an internal control to normalize transcript levels. The primer details are shown in Supplementary Table 3. A cycling temperature of 57 °C and the criterion of a single peak on the melting curve were used to confirm the specificity of designed primer pairs.
Data Availability
All data generated or analysed during this study are included in this published article and its Supplementary Information files.
References
Berger, F., Grini, P. E. & Schnittger, A. Endosperm: an integrator of seed growth and development. Curr Opin Plant Biol 9, 664–670, https://doi.org/10.1016/j.pbi.2006.09.015 (2006).
Sun, X. D., Shantharaj, D., Kang, X. J. & Ni, M. Transcriptional and hormonal signaling control of Arabidopsis seed development. Curr Opin Plant Biol 13, 611–620, https://doi.org/10.1016/j.pbi.2010.08.009 (2010).
Kesavan, M., Song, J. T. & Seo, H. S. Seed size: a priority trait in cereal crops. Physiol Plantarum 147, 113–120, https://doi.org/10.1111/j.1399-3054.2012.01664.x (2013).
Li, N. & Li, Y. H. Maternal control of seed size in plants. J Exp Bot 66, 1087–1097, https://doi.org/10.1093/jxb/eru549 (2015).
Sundaresan, V. Control of seed size in plants. Proc Natl Acad Sci USA 102, 17887–17888, https://doi.org/10.1073/pnas.0509021102 (2005).
Yan, S. et al. Seed size is determined by the combinations of the genes controlling different seed characteristics in rice. Theor Appl Genet 123, 1173–1181, https://doi.org/10.1007/s00122-011-1657-x (2011).
YongJae, K. et al. Development of near isogenic lines with various seed sizes and study on seed size-related characteristics in watermelon. Korean Journal of Breeding Science 41, 403–411 (2009).
Weetman, L. M. Inheritance and correlation of shape, size and color in the watermelon, Citrullus vulgaris Schrad. Iowa Agr. Expt. Sta. Res. Bul. 228, 222–256 (1937).
Poole, C. F., Grimball, P. C. & Porter, D. R. Inheritance of seed characters in watermelon. Jour Agr Res 66, 433–456 (1941).
Kensler, T. & Barham, W. The inheritance of seed size in watermelon. Proc Amer Soc Hort Sci. 480–484 (1958).
Shimotsuma, M. Cytogenetical studies in the Genus Citrullus: VII. inheritance of several characters in watermelons. Jap J Breeding 13, 235–240 (1963).
Tanaka, T., Wimol, S. & Mizutani, T. Inheritance of fruit shape and seed size of watermelon [Citrullus lanatus]. J Japan Soc Hort Sci 64, 543–548 (1995).
Zhang, X., Rhodes, B. & Wang, M. Genes controlling watermelon seed size, Cucurbitaceae. 144–147 (1994).
Zhang, J. Inheritance of seed size from diverse crosses in watermelon. Cucurbit Genetics Coop Rpt 19, 67–69 (1996).
Hawkins, L. K., Dane, F. & Kubisiak, T. L. Molecular markers associated with morphological traits in watermelon. Hortscience 36, 1318–1322 (2001).
Prothro, J. et al. Main and epistatic quantitative trait loci associated with seed size in watermelon. J Am Soc Hortic Sci 137, 452–457 (2012).
Shang, Y. et al. Plant science. Biosynthesis, regulation, and domestication of bitterness in cucumber. Science 346, 1084–1088, https://doi.org/10.1126/science.1259215 (2014).
Meru, G. & McGregor, C. Genetic mapping of seed traits correlated with seed oil percentage in watermelon. Hortscience 48, 955–959 (2013).
Kim, K. H. et al. Major quantitative trait loci and putative candidate genes for powdery mildew resistance and fruit-related traits revealed by an Intraspecific genetic map for watermelon (Citrullus lanatus var. lanatus). PLoS One 10, https://doi.org/10.1371/journal.pone.0145665 (2015).
Shang, J. et al. Construction of a high-density genetic map for watermelon (Citrullus lanatus L.) based on large-scale SNP discovery by specific length amplified fragment sequencing (SLAF-seq). Scientia Horticulturae 203, 38–46, https://doi.org/10.1016/j.scienta.2016.03.007 (2016).
Wang, L. et al. Construction of a high-density genetic linkage map in pear (Pyrus communis x Pyrus pyrifolia nakai) using SSRs and SNPs developed by SLAF-seq. Sci Hortic-Amsterdam 218, 198–204, https://doi.org/10.1016/j.scienta.2017.02.015 (2017).
Guo, S. et al. The draft genome of watermelon (Citrullus lanatus) and resequencing of 20 diverse accessions. Nat Genet 45, 51–58, https://doi.org/10.1038/ng.2470 (2013).
Shi, J. Q. et al. Genome-wide microsatellite characterization and marker development in the sequenced Brassica crop species. DNA Research 21, 53–68, https://doi.org/10.1093/dnares/dst040 (2014).
Serrano, M., Parra, S., Alcaraz, L. D. & Guzman, P. The ATL gene family from Arabidopsis thaliana and Oryza sativa comprises a large number of putative ubiquitin ligases of the RING-H2 type. J Mol Evol 62, 434–445, https://doi.org/10.1007/s00239-005-0038-y (2006).
He, Y. H. & Gan, S. S. A novel zinc-finger protein with a proline-rich domain mediates ABA-regulated seed dormancy in Arabidopsis. Plant Mol Biol 54, 1–9, https://doi.org/10.1023/B:Plan.0000028730.10834.E3 (2004).
Kitagawa, K. et al. A novel kinesin 13 protein regulating rice seed length. Plant Cell Physiol 51, 1315–1329, https://doi.org/10.1093/pcp/pcq092 (2010).
Zhang, Y., Luo, L., Liu, T., Xu, C. & Xing, Y. Four rice QTL controlling number of spikelets per panicle expressed the characteristics of single Mendelian gene in near isogenic backgrounds. Theor Appl Genet 118, 1035–1044 (2009).
Fujino, K. et al. Molecular identification of a major quantitative trait locus, qLTG3-1, controlling low-temperature germinability in rice. Proc Natl Acad Sci USA 105, 12623–12628, https://doi.org/10.1073/pnas.0805303105 (2008).
Wan, X. Y. et al. QTL analysis for rice grain length and fine mapping of an identified QTL with stable and major effects. Theor Appl Genet 112, 1258–1270, https://doi.org/10.1007/s00122-006-0227-0 (2006).
Eshed, Y. & Zamir, D. A genomic library of Lycopersicon pennellii in L. esculentum: A tool for fine mapping of genes. Euphytica 79, 175–179 (1994).
Salvi, S. & Tuberosa, R. To clone or not to clone plant QTLs: present and future challenges. Trends Plant Sci 10, 297–304 (2005).
Noh, B., Murphy, A. S. & Spalding, E. P. Multidrug resistance-like genes of Arabidopsis required for auxin transport and auxin-mediated development. Plant Cell 13, 2441–2454, https://doi.org/10.1105/tpc.13.11.2441 (2001).
Noh, B., Bandyopadhyay, A., Peer, W. A., Spalding, E. P. & Murphy, A. S. Enhanced gravi- and phototropism in plant mdr mutants mislocalizing the auxin efflux protein PIN1. Nature 423, 999–1002, https://doi.org/10.1038/nature01716 (2003).
Goodman, C. D., Casati, P. & Walbot, V. A multidrug resistance-associated protein involved in anthocyanin transport in Zea mays. Plant Cell 16, 1812–1826, https://doi.org/10.1105/tpc.022574 (2004).
Zhang, H. W. et al. A DTX/MATE-Type transporter facilitates abscisic acid efflux and modulates ABA sensitivity and drought tolerance in Arabidopsis. Mol Plant 7, 1522–1532, https://doi.org/10.1093/mp/ssu063 (2014).
Debeaujon, I., Peeters, A. J. M., Leon-Kloosterziel, K. M. & Koornneef, M. The TRANSPARENT TESTA12 gene of Arabidopsis encodes a multidrug secondary transporter-like protein required for flavonoid sequestration in vacuoles of the seed coat endothelium. Plant Cell 13, 853–871, https://doi.org/10.1105/Tpc.13.4.853 (2001).
Wang, R. et al. A subgroup of MATE transporter genes regulates hypocotyl cell elongation in. Arabidopsis. J Exp Bot 66, 6327–6343, https://doi.org/10.1093/jxb/erv344 (2015).
Schmitt, A. O., Assmus, J., Bortfeldt, R. H. & Brockmann, G. A. CandiSNPer: a web tool for the identification of candidate SNPs for causal variants. Bioinformatics 26, 969–970, https://doi.org/10.1093/bioinformatics/btq068 (2010).
Institute, S. A. S. SAS/STAT user’s guide, version 8. Vol. 2 (SAS Institute, 1999).
Zeng, Z.-B. Precision mapping of quantitative trait loci. Genetics 136, 1457–1468 (1994).
Churchill, G. A. & Doerge, R. W. Empirical threshold values for quantitative trait mapping. Genetics 138, 963–971 (1994).
Wang, S. B. et al. Mapping small-effect and linked quantitative trait loci for complex traits in backcross or DH populations via a multi-locus GWAS methodology. Sci Rep 6, 29951, https://doi.org/10.1038/srep29951 (2016).
Jiang, C. & Zeng, Z. B. Multiple trait analysis of genetic mapping for quantitative trait loci. Genetics 140, 1111–1127 (1995).
Zhu, J. Analysis of conditional genetic-effects and variance-components in developmental genetics. Genetics 141, 1633–1639 (1995).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760, https://doi.org/10.1093/bioinformatics/btp324 (2009).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
McKenna, A. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20, 1297–1303, https://doi.org/10.1101/gr.107524.110 (2010).
Neff, M. M., Turk, E. & Kalishman, M. Web-based primer design for single nucleotide polymorphism analysis. Trends Genet 18, 613–615 (2002).
Clarke, K. R. & Warwick, R. M. PRIMER v5: user manual/tutorial. (Primer-E Limited, 2001).
Rychlik, W. OLIGO 7 primer analysis software. (Springer, 2007).
Doyle, J. J. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem bull 19, 11–15 (1987).
Kosambi, D. D. The estimation of map distances from recombination values. Annals of Eugenics 12, 172–175 (1943).
Acknowledgements
This research was supported by the National Nature Science Foundation of China (31601779), the National Key R&D Program of China (2016YFD0100204-26), the Central Public-interest Scientific Institution Basal Research Fund (1610192016207 and 1610192018108), the Special Protection and Utilization of the Crop Germplasm Resources of China (2014NWB038), the National R&D Infrastructure and Facility Development Program of China and the Agricultural Science and Technology Innovation Program of China (CAAS-ASTIP-2017-ZFRI). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. We also thank the research sharing service platform of Zhengzhou Fruit Research Institute, Chinese Academy of Agricultural Sciences for providing the instruments and technologies.
Author information
Authors and Affiliations
Contributions
Conceived and designed the experiments: N.L., S.W.M.; performed the experiments: N.L., J.L.S., D.Z., N.N.L.; analysed the data: N.L.; contributed reagents/materials/analytic tools: N.L., J.L.S., J.M.W., S.W.M.; wrote the manuscript: N.L. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, N., Shang, J., Wang, J. et al. Fine mapping and discovery of candidate genes for seed size in watermelon by genome survey sequencing. Sci Rep 8, 17843 (2018). https://doi.org/10.1038/s41598-018-36104-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-36104-w
This article is cited by
-
A nonsynonymous mutation in BhLS, encoding an acyl-CoA N-acyltransferase leads to fruit and seed size variation in wax gourd (Benincasa hispida)
Theoretical and Applied Genetics (2024)
-
Meta-analysis of qualitative and quantitative trait variation in sweet watermelon and citron watermelon genetic resources
Genetic Resources and Crop Evolution (2023)
-
Integrated analysis of biparental and natural populations reveals CRIB domain-containing protein underlying seed coat crack trait in watermelon
Theoretical and Applied Genetics (2023)
-
CAPS marker-base genetic linkage mapping and QTL analysis for watermelon ovary, fruit and seed-related traits
Euphytica (2022)
-
Genetic mapping of loci determining seed coat color and size in watermelon
Euphytica (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
