
Abstract
Infections caused by metallo-beta-lactamase-producing organisms (MBLs) are a global health threat. Our understanding of transmission dynamics and how MBLs establish endemicity remains limited. We analysed two decades of blaIMP-4 evolution in a hospital using sequence data from 270 clinical and environmental isolates (including 169 completed genomes) and identified the blaIMP-4 gene across 7 Gram-negative genera, 68 bacterial strains and 7 distinct plasmid types. We showed how an initial multi-species outbreak of conserved IncC plasmids (95 genomes across 37 strains) allowed endemicity to be established through the ability of blaIMP-4 to disseminate in successful strain-genetic setting pairs we termed propagators, in particular Serratia marcescens and Enterobacter hormaechei. From this reservoir, blaIMP-4 persisted through diversification of genetic settings that resulted from transfer of blaIMP-4 plasmids between bacterial hosts and of the integron carrying blaIMP-4 between plasmids. Our findings provide a framework for understanding endemicity and spread of MBLs and may have broader applicability to other carbapenemase-producing organisms.
Similar content being viewed by others


Introduction
Carbapenemase-producing organisms (CPOs) are now endemic in many regions. While there has been a significant focus on blaKPC due to its spread through North America and Europe, metallo-beta-lactamases (MBLs) (e.g. blaNDM, blaIMP and blaVIM) are endemic through much of Asia and Oceania including Australia, where blaIMP carbapenemases have dominated1,2,3,4,5,6,7,8,9. Treatment options for infections caused by CPOs, particularly MBL-harbouring organisms, remain severely limited, highlighting the need to stop further spread of these extensively drug-resistant organisms. The mechanisms for carbapenemase spread differ according to carbapenemase type. Some carbapenemases spread through close associations with successful strains or lineages (e.g. blaKPC-2/3 and Klebsiella pneumoniae clonal complex 258), while for others, spread is mediated through association with specific plasmids (e.g. blaOXA-48 and broad-host range IncL plasmids)10,11. Notably, MBLs spread through both lineage-related clonal expansion and diverse plasmid types12,13. While surveillance studies have captured some of these data, there have been few efforts to assess how these mechanisms of spread evolve over time. Understanding the transmission dynamics of carbapenem resistance genes will be crucial to inform future infection prevention efforts.
Previous work from our group and others have identified that the spread of MBLs, and particularly blaIMP-4, is often driven by dissemination by transposons of a class 1 integron that has been able to insert into several genetic settings (henceforth defined as either chromosomal integration or different plasmid types carrying blaIMP-4)2,4,6,9,14,15. Furthermore, the gene cassettes (such as that carrying blaIMP-4) may also be a source of dissemination by being able to enter different class 1 integrons16. The ability to study horizontal gene transfer has been significantly advanced by long-read sequencing, which enables high-quality de novo assembly of bacterial genomes, including highly repetitive regions such as plasmids. Utilising long-read sequencing to generate completed, closed bacterial genomes provides a unique opportunity to study the complex, multi-level (bacterial strain, plasmid, gene) transmission dynamics that are likely occurring during MBL spread. When combined with short-read sequencing data, an unprecedented level of detail of the genetic context and likely mechanisms of an outbreak or endemicity is possible.
MBL-producing Gram-negative bacteria, dominated by blaIMP-4, have been isolated in our institution (The Alfred Hospital) since 20025,6,7,15. After an initial outbreak period in 2004–2005, we experienced hyperendemicity, with a repeated outbreak period from 2017–2020. We aimed to assess the genetic settings of blaIMP-4, its evolution over time, and the transmission pathways that resulted in repeated outbreaks and endemicity.
In this work we used long- and short-read whole genome sequencing to characterise the genetic settings of blaIMP-4 in bacterial chromosomes and plasmids from 277 clinical and environmental isolates from 2002–2020. This allowed us to track the spread of blaIMP-4 in 7 plasmid types and multiple chromosomal settings. In the 18-year period, we noted incredible plasticity of blaIMP-4 persistence, with vertical spread through the transmission of dominant strains and horizontal spread of both plasmids and the class 1 integron carrying blaIMP-4. We also identified a persistent reservoir of blaIMP-4 in IncC plasmids in both clinical and environmental isolates. Our findings highlight the need for integration of long-read sequencing into CPO surveillance, as well as for multi-modal infection prevention approaches that address the diverse forms of CPO spread.
Results
bla IMP-4 found in diverse clinical and environmental isolates spanning two decades
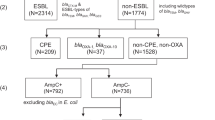
We sequenced 277 blaIMP-4-harbouring isolates from an institutional collection of carbapenem-resistant isolates systematically collected from 2002–2020, including 264 clinical isolates from 196 patients and 13 environmental isolates (Supplementary Dataset 1). This included short-read (Illumina) data on all isolates and long-read (Oxford Nanopore) data on 172 isolates that best represented the strains across the study time periods. Seven isolates failed quality control and were excluded. In total, we analysed 270 isolates that were made up of 68 bacterial strains (defined as unique species/multi-locus sequence type [MLST] combinations) from 7 Gram-negative genera, highlighting the diversity of bacterial hosts for blaIMP-4 (Fig. 1a). The five most frequent strains accounted for 190/270 (70%) genomes and included Serratia marcescens (52/270 isolates, 19%), Enterobacter hormaechei ST190 (44/270 genomes, 17%), E. hormaechei ST93 (36/270 genomes, 13%), Pseudomonas aeruginosa ST111 (35/270 genomes, 13%) and E. hormaechei ST114 (23/270 genomes, 9%) (Fig. 1a). In addition to blaIMP-4, 8/270 (3%) genomes carried other carbapenemase genes (4 blaOXA-58, 2 blaNDM-7, 1 blaNDM-1, 1 blaKPC-2, 1 blaOXA-500) and 121/270 (44%) carried mcr-9.1, a novel determinant of colistin resistance14 (Supplementary Dataset 1).

a Pie chart showing key bacterial host strains of blaIMP-4. blaIMP-4 was noted in 7 bacterial genera and 68 bacterial host strains. b Genetic settings and bacterial host species of blaIMP-4 over course of study, as defined by three distinct periods. Abbreviations: No number, Lin lineage, ST sequence type.
bla IMP-4 detected in multiple plasmid and chromosomal genetic settings across three distinct time periods
We first determined the genetic setting of blaIMP-4 using 169 completed, circularised genomes (three non-circularised genomes were excluded). blaIMP-4-carrying plasmids were clustered using MOB-typer17, which uses a whole-sequence-based typing system to provide cluster codes for reconstruction and tracking of plasmids. Representative plasmids from each cluster were then used as references for mapping of the 99 genomes with short-read data only and the three non-circularised genomes (Supplementary Dataset 1). Overall, 230 and 40 isolates carried blaIMP-4 on a plasmid or on the chromosome, respectively, with seven distinct plasmid types identified and chromosomal integration in multiple strains (Fig. 1b and Supp. Table 1). No genomes showed concurrent chromosomal integration and plasmid carriage of blaIMP-4. For the majority of isolates (151/169, 89%), blaIMP-4 was situated in a class 1 integron most commonly comprising the blaIMP-4-qacG-aacA4-catB3-qacE-sul1 cassette array. The bacterial host strain-plasmid relationships evolved over the course of the study, with three distinct time periods (Fig. 1b).
Outbreak initiation and establishment of bla IMP-4 endemicity
blaIMP-4 was first noted in a clinical S. marcescens isolate in 2002, with blaIMP-4 being carried on an IncC plasmid (Supp. Fig. 1). It took ~2 years before further blaIMP-4-carrying Gram-negative bacteria were identified, and these were dominated by an IncC genetic setting or chromosomal blaIMP-4 in P. aeruginosa (Fig. 1b). Notably, the first S. marcescens lineage (here called lineage 1) with IncC-carrying blaIMP-4 became a successful lineage across the entire study period (Fig. 1b). S. marcescens lineage 1 included 47 genomes from both clinical and environmental (intensive care unit [ICU] sinks) isolates. Based on their genetic relatedness (median pairwise single nucleotide variant [SNV] distance of 8 [IQR 4–11]), vertical transmission was most likely (Fig. 2a and Supp. Table 2). The IncC plasmid was rapidly noted in diverse bacterial hosts, with expansion from S. marcescens into 13 other bacterial strains in 2004 alone. Ultimately, this plasmid was noted in 95 genomes across 37 strains (Fig. 2b), with S. marcescens genomes accounting for the majority (52/95 [54.7%]). Despite the diversity of IncC-carrying bacterial hosts over the 18 years, IncC plasmids were highly conserved, with only 11 SNVs across the plasmid backbone (median pairwise distance of 0 SNVs) (Supp. Table 3) and almost all (70/71, 98.5%) IncC plasmids belonged to the same MOB-typer primary/secondary clusters (AA860 AJ266, Supp. Table 1). We noted seven IncC mosaic plasmids with additional replicon types that were excluded from further clustering analyses (Supp. Table 4).

a Phylogenetic analysis of blaIMP-4-carrying S. marcescens genomes from Alfred Hospital. Inset panel locates S. marcescens genomes from the Alfred Hospital in a global S. marcescens phylogeny. Outer panel shows Alfred Hospital genomes only, indicating that they formed two distinct lineages and were detected both in clinical and environmental samples. b Phylogenetic analysis of blaIMP-4 IncC plasmids using Mashtree. IncC plasmids entered 37 bacterial host strains but remained stable with four flanking region clusters and one integron SNV profile accounting for 93% and 90% plasmids, respectively. c Analysis of blaIMP-4 flanking regions in P. aeruginosa ST111. Three flanking clusters were detected in P. aeruginosa ST111. Differences are shown to the class 1 integron present in contemporaneous blaIMP-4 IncC plasmids (top), with absence of catB3 cassette in flanking cluster 1 (in genomes from 2004–2012), absence of qacG, aac4 and catB3 cassettes in flanking cluster 2 (2014–2015) and distinct flanking regions and absence of catB3 cassette in flanking cluster 3 (2018). Abbreviations: bp base pairs, Lin. lineage, SNV single nucleotide variant, ST sequence type.
We used Mashtree18 to further characterise relatedness of the 64 non-mosaic IncC plasmids, which highlighted the similarity of plasmids across bacterial hosts and different time periods (Fig. 2b). This was also reflected in the blaIMP-4 flanking regions and integron sequences. Cluster analysis of the flanking regions up to 5000 bp upstream and downstream of blaIMP-4 using Flanker19 identified only four flanking regions (excluding singletons) in IncC plasmids, accounting for 67/71 (94%) genomes (Fig. 2b). The blaIMP-4 containing integrons in the IncC plasmids were also highly similar, with a single SNV profile (TGGTCGACGCCT) accounting for 63/69 (91%) plasmids with intact integrons (Fig. 2b). Taken together, these findings suggest that blaIMP-4 containing IncC plasmids dominated the early outbreak period and established endemicity through their ability to rapidly spread across different bacterial hosts while maintaining stability. S. marcescens was a persistent host and reservoir for blaIMP-4 IncC during this time period (2002–2010) (Supp. Fig. 1).
In addition to blaIMP-4 IncC plasmids, chromosomal integration of blaIMP-4 into P. aeruginosa ST111 (a global MDR lineage)20 was also a dominant feature of this early time period (Fig. 1b and Supp. Fig. 1). These isolates were rapidly noted in nine patients in 2004 and continued to be isolated until 2018. The pseudomonal isolates were highly related (mean pairwise SNV distance of 1.6 SNVs vs 38.7 SNVs between Alfred and publicly available ST111 genomes [P < 0.001]) (Supp. Fig. 2a) but the blaIMP-4 containing integron and the flanking regions differed depending on the time period of isolation, and were also different to the integron and flanking region sequence of the IncC plasmids (Fig. 2c). Despite being temporally associated, the chromosomal integration of blaIMP-4 in P. aeruginosa ST111 with a different integron structure and flanking regions suggests that blaIMP-4 entry into P. aeruginosa likely arose independently of the blaIMP-4 IncC plasmids.
Low endemicity of bla IMP-4 and entry into novel plasmids
Apart from ongoing isolation of blaIMP-4 IncC plasmids (predominately in S. marcescens) and chromosomal blaIMP-4 P. aeruginosa, the evolution of the following time period (2011–2015) was characterised by blaIMP-4 entering novel plasmids in E. hormaechei ST114 (a global MDR nosocomial Enterobacter clone)21 (Fig. 1b and Supp. Fig. 1). At the start of the outbreak, there were a small number of E. hormaechei ST114 with blaIMP-4-carrying IncC plasmids, but during this period, E. hormaechei ST114 acquired blaIMP-4-carrying IncFIB and IncFIA/IncFIB/IncP plasmids (Fig. 1b). Phylogenomic analysis showed that the E. hormaechei ST114 isolates were more diverse (median pairwise SNV distance 35, IQR 27–46) but the Alfred Hospital isolates still clustered more closely than other publicly available genomes (Fig. 3a and Supp. Table 2). To determine if the IncC plasmids were the source of blaIMP-4 in the IncFIB and IncFIA/IncFIB/IncP plasmids, we analysed the blaIMP-4 flanking regions across the three plasmids (Fig. 3b). We noted homology of the 3850 bp upstream and 305 bp downstream regions of the integron in IncC, IncFIA/IncFIB/IncP and IncFIB plasmids (clusters A, B and C) with Tn3 transposons and DNA recombinases (hin) located immediately upstream. (Fig. 3b). These three plasmids also shared the same integron SNV profile (TGGTCGACGCCT) (Supp. Fig. 3). Taken together, these findings suggested that as the blaIMP-4-harbouring IncC plasmids became endemic during the first time period (2002–2010), the outbreak evolved whereby the IncC plasmids served as a blaIMP-4 reservoir not only for inter-strain plasmid transfer, but also transfer of the blaIMP-4 integron and flanking regions between IncC, IncFIA/IncFIB/IncP and IncFIB plasmids.

a Phylogenetic analysis of blaIMP-4-carrying E. hormaechei ST114 genomes from Alfred Hospital. Inset panel places E. hormaechei ST114 genomes from the Alfred Hospital in a global E. hormaechei ST114 phylogeny. Outer panel shows Alfred Hospital genomes only. Although clonally related, they were more diverse than other key blaIMP-4 bacterial host strains and were able to act as versatile acceptors of blaIMP-4 in different genetic settings, including four different plasmid types throughout the study. b Clustering analysis of flanking regions 5000 bp upstream and downstream of blaIMP-4 using Flanker. The six key flanking region clusters with >5 genomes are shown on the left and the genetic context is shown on the right. IncFIB and IncFIA/IncFIB/IncP plasmids clustered with IncC plasmids throughout the analysis and shared homology both upstream and downstream of blaIMP-4. Abbreviations: bp. base pairs, SNV single nucleotide variant.
Hyperendemicity and repeat outbreaks driven by clonal expansion of new bacterial strains and inter-species plasmid spread
The most recent time period (2016–2020) was characterised by complex, multi-level transmission dynamics resulting from the emergence of several new and highly successful blaIMP-4 plasmids and clonal expansion of E. hormaechei ST190 and ST93 host strains (Fig. 1b). Ongoing circulation of blaIMP-4 in genetic settings and bacterial strains from prior periods was also observed (Fig. 1b). Early in this period, blaIMP-4 was identified in a new plasmid, IncL/M, first in E. hormaechei ST114, which was its fourth blaIMP-4 carrying plasmid, and then in a wide range of other bacterial strains (n = 16) (Fig. 1b). All IncL/M plasmids belonged to the same MOB-typer cluster, shared the same SNV profile in the integron (GGGTCGACGCCT) and 14/17 shared the same flanking cluster (Cluster G) (Fig. 3b). The three plasmids with other flanking clusters had minor variations in the Cluster G flanking region leading to them being clustered as singletons. These flanking regions were distinct from all other blaIMP-4 plasmids, however the same integron SNV profile was also noted in other plasmids (Supp. Table 5 and Supp. Fig. 3).
In 2017, blaIMP-4 was detected for the first time in IncHI2A plasmids in a small outbreak of Klebsiella oxytoca ST278 and Klebsiella michiganensis ST50 (Fig. 1b). The first IncHI2A plasmid (type 1—as defined by MOB-typer cluster AA739 AJ055) then spread to E. hormaechei ST190 and a second IncHI2A plasmid (type 2—MOB-typer cluster AA739 AJ058) emerged in E. hormaechei ST93, with both bacterial strains undergoing significant clonal expansion and contributing to a repeated outbreak and hyperendemicity from 2017–2020 (Figs. 1b and 4a). Bacterial isolates carrying blaIMP-4 on these two IncHI2A plasmids ultimately accounted for 98/161 (61%) of the sequenced genomes in that period (with 36/98 E. hormaechei ST93 and 43/98 E. hormaechei ST190) (Fig. 4a). The E. hormaechei ST93 and ST190 bacterial hosts were highly clonal with a median pairwise SNV distance of 9 (IQR 2-14) and 3 (IQR 2–4), respectively (Supp. Fig. 2b and 2c, Supp. Table 2). In addition to these two strains, the IncHI2A plasmids were found in 13 other strains (Fig. 4a). Analysis of the blaIMP-4 flanking regions and integrons in the IncHI2A plasmids showed the same flanking sequence (Cluster F) across 36/47 (77%) plasmids and the same integron SNV profile (GGGTCGACGTCT) in 35/47 (74%) plasmids across both IncHI2A plasmid types (Fig. 4a). These flanking sequences and integron SNV profiles were not found in other plasmid types (Supp. Fig. 3), suggesting that they may have arisen independently of other blaIMP-4 genetic settings.

a Phylogenetic analysis of IncHI2A plasmids using Mashtree. Two distinct plasmid types (IncHI2A type 1 and type 2) were noted from 2016–2020 and rapidly entered 15 bacterial strains. These plasmids shared flanking regions and integron SNV profiles, which were distinct to those noted in other plasmid types. b, c Comparative analysis of blaIMP-4-carrying IncHI2A plasmids to non-blaIMP-4 IncHI2A plasmids from E. hormaechei ST190, ST93 and ST114 from Alfred Hospital. IncHI2A type 1 plasmids from E. hormaechei ST190 had homology over 100% of the non-blaIMP-4 plasmid, with the addition of a 33 kbp region carrying the class 1 integron in the blaIMP-4 plasmid. IncHI2A type 2 plasmids from E. hormaechei ST93 shared homology across 97.6% of the non-blaIMP-4 plasmid, with the blaIMP-4 integron contained in a mosaic region. Abbreviations: bp base pairs, SNV single nucleotide variant, ST sequence type.
To understand the rapid appearance of blaIMP-4 IncHI2A plasmids, we compared them to carbapenem-susceptible, non-blaIMP-4 IncHI2A plasmids in single E. hormaechei ST114, ST190 and ST93 genomes from our institution. The plasmids were highly similar between blaIMP-4 and non-blaIMP-4 bacterial strains (Fig. 4b). The IncHI2A type 1 plasmids from blaIMP-4 E. hormaechei ST190 had the addition of a 33 kbp region carrying the class 1 integron with blaIMP-4 (Fig. 4b). The IncHI2A type 2 plasmids from blaIMP-4 E. hormaechei ST93 shared 97.6% homology with the non-blaIMP-4 plasmid, with the blaIMP-4 integron contained in a mosaic region (Fig. 4b, c). These data suggested integration of blaIMP-4 into pre-existing carbapenem-susceptible Enterobacter carrying IncHI2A plasmids, with mobilisation of the regions upstream and downstream. These flanking regions in IncHI2A plasmids were distinct to those in other blaIMP-4 plasmids, with a different IS110-like insertion sequence/Tn3 transposon combination (Fig. 4c).
While the arrival of new IncHI2A and IncL/M plasmids was the major contributor to the repeated outbreak and hyperendemicity during this period, we saw ongoing circulation of blaIMP-4 in prior genetic settings. In particular, we saw IncC plasmids circulating (32/161 genomes, 19.9%), including in a novel lineage of S. marcescens in 2019 (lineage 2—Fig. 2a). We also noted blaIMP-4-harbouring P. aeruginosa ST111 (1 genome) and E. hormaechei ST114 with blaIMP-4 IncFIA/IncFIB/IncP plasmids (9 genomes). This reflected a cumulative trend where the prior blaIMP-4 genetic settings persisted in the context of new bacterial strains and plasmids, rather than waxing and waning over time.
An environmental reservoir of bla IMP-4 plasmids
In response to the outbreak in the most recent time period, we conducted environmental screening of ICU sinks from 2019–2020 and cultured 34 blaIMP-4 isolates (33 S. marcescens and one E. hormaechei), with 11 isolates selected for sequencing. Despite E. hormaechei predominating in blaIMP-4 clinical isolates, 6 genomes were S. marcescens lineage 1, 4 S. marcescens lineage 2, and one E. hormaechei ST190. These genomes closely matched clinical isolates, with blaIMP-4 being located on IncC plasmids and IncHI2A type 1 plasmids in S. marcescens and E. hormaechei ST190, respectively (Figs. 2b and 4a). This indicated that sinks were a possible reservoir for blaIMP-4 and may have explained the persistence of S. marcescens with closely matching IncC plasmids throughout the study (Fig. 2b).
Within-patient and between-patient bla IMP-4 analyses show importance of diversification of genetic settings through plasmid transfer
Patients with serial blaIMP-4-harbouring genomes available followed different trajectories of blaIMP-4 carriage (Fig. 5a). Multiple colonisation events were noted in 5/41 patients, with blaIMP-4 being located in distinct genetic settings (i.e. differing plasmids and/or chromosomal integration). Flanking region/integron SNV profiles also differed, making within-patient integron transfer unlikely. Possible within-patient inter-strain transfer of key blaIMP-4 plasmids (IncC, IncL/M, IncHI2A types 1 and 2) occurred in 10/41 patients with the same blaIMP-4 plasmid types being noted in multiple strains (Fig. 5a). In 7 patients, the evidence was particularly compelling as plasmids had identical flanking sequences and integron SNV profiles across different bacterial hosts. Persisting colonisation was noted in 26 patients, with the same strain and same blaIMP-4 genetic setting repeatedly isolated.

a Longitudinal analysis of blaIMP-4 in patients with multiple genomes available. Each line represents a patient with colours representing the bacterial host and shapes representing the blaIMP-4 genetic setting. b Analysis of potential transmission events between patients. Individual patients are shown as vertices. Edges were drawn if there was spatiotemporal overlap on a hospital wards (indicated by edge colour) and a genomic criterion was met. In the left panel, the genomic criterion was having the same blaIMP-4 bacterial strain with SNV distance from phylogenetic analysis indicated by shading. In the right panel, the genomic criterion incorporated both the strain transmission analysis as well as detecting presence of blaIMP-4 in the same plasmid (as defined by MOB-typer cluster) in different bacterial strains. The additional benefit of plasmid analysis is indicated by the blue shading of the potential transmission events in this panel. The red shading indicates the patient with the highest betweenness-centrality and their contacts. Abbreviations: ICU intensive care unit, Lin. lineage, SNV single nucleotide variant, ST sequence type.
We then used patient movement data (available in 127 patients from 2013 onwards) to establish putative transmission events (Fig. 5b), defined as spatiotemporal overlap between patients and genomic evidence of potential transmission. For genomic evidence, we considered both strain transmission (same bacterial host strain carrying blaIMP-4 in the same genetic setting) and plasmid transmission (detection of the same plasmid by MOB-typer cluster in different bacterial strains). We linked 71/127 (56%) patients using these definitions and identified the ICU as a major transmission site with 36/76 (47%) potential transmission events across 7/16 (44%) transmission networks, including the two largest networks (23 and 9 patients, respectively). While strain transmission contributed significantly, use of long-read sequencing to detect potential plasmid transmission allowed us to detect an additional 5/10 (50%) wards, 7/16 (44%) transmission networks and link 22/71 (31%) patients beyond what was identified for strain transmission alone (Fig. 5b). We measured betweenness centrality to identify key patients involved in transmission22. The patient with the highest betweenness centrality (164.0 vs mean 4.9) had a > 4-month ICU admission and was implicated in 7 transmission events, placing them at the centre of the large 23-patient network spanning those two strains and plasmid types (E. hormaechei ST190 with IncHI2A type 1 plasmid and E. hormaechei ST93 with IncHI2A type 2 plasmid) (Fig. 5b). A further transmission event to a single patient then occurred during an admission on the Cardiology ward 6 months later.
Discussion
The spread of carbapenemases is the major driver of carbapenem resistance globally23 and has been the focus of numerous cross-sectional studies12,13,24. To date, there have been limited efforts to study carbapenem resistance over extended time periods25,26. In this study we had a unique opportunity to analyse two decades of blaIMP-4 carbapenemases in our institution and gained important insights into how blaIMP-4 caused outbreaks and perpetuated endemicity. blaIMP-4 spread occurred through multiple mechanisms including strain transmission, plasmid transmission and transfer of the blaIMP-4 class 1 integron. Each of these had a different qualitative and quantitative contribution to blaIMP-4 persisting at our institution, highlighting that endemicity is a nuanced process requiring these mechanisms to act in concert. These findings carry important implications for prevention of future carbapenemase endemicity.
From a pathogen perspective, we propose that there are two key conditions required for blaIMP-4 endemicity. Firstly, there is a need for diversification of genetic settings for the resistance determinant, which in our study occurred through extensive inter-strain transmission of key blaIMP-4 plasmids (IncC, IncHIA2 type 1 and type 2, IncL/M), as well as mobilisation by transposons of the class I integron and entry into new plasmids (IncFIB, IncFIA/IncFIB/IncP). This ability to diversify led to the initial establishment of endemicity with IncC plasmids, and also to the period of hyperendemicity due to the emergence of a novel context in IncHI2A and IncL/M plasmids. To study this emergence, we demonstrated that blaIMP-4 IncHI2A plasmids were highly similar to non-blaIMP-4 IncHI2A plasmids in E. hormaechei ST93, ST114 and ST190 that may have served as acceptors for the blaIMP-4 integron. In addition, there was possible importation from outside sources: blaIMP-4 has been found in both IncHI2A and IncL/M plasmids in Australian isolates2,4,9,27 and blaIMP-4-harbouring IncHI2A plasmids are emerging as a global issue, having been noted in a recent multi-hospital outbreak in the United Kingdom28.
The second condition is propagation of blaIMP-4 through the establishment of high-risk strain-genetic setting pairs we term propagators. While we noted blaIMP-4 in 68 strains during the study, five strains accounted for 190/270 (70%) genomes and 140/196 (71%) patients colonised with blaIMP-4. The first of these was S. marcescens lineage 1 (IncC plasmids), which defined the early period of the study and continued to persist throughout. This propagator pair was able to act as a reservoir of blaIMP-4, likely through occupying an environmental niche as we noted during sampling of ICU sinks. The colonisation of hospital plumbing by CPOs has been well documented9,29,30,31,32, including blaIMP-4-harbouring S. marcescens in an Australian setting that was unable to be eradicated33. This environmental colonisation probably enabled clonal spread of blaIMP-4 -harbouring S. marcescens and may have driven diversification through inter-strain transfer of blaIMP-4 IncC plasmids and inter-plasmid transfer of the blaIMP-4 class I integron. Other propagators emerged at various junctures including P. aeruginosa ST111 (chromosome) and E. hormaechei ST114 (IncFIA/IncFIB/IncP plasmids), then E. hormaechei ST190 and ST93 (IncHI2A type 1 and type 2 plasmids, respectively). Clonal spread of propagators was therefore central to establishing and maintaining blaIMP-4 endemicity, as well as leading to a repeated outbreak and hyperendemicity in the final period of the study. These findings broadly fit the ‘multiple lineages, multiple plasmids’ designation proposed by David et al. when analysing carbapenemase spread in K. pneumoniae12 but we demonstrated that the dynamics of carbapenemase endemicity in our setting were significantly more complex with clonal transmission of propagator strains, inter-strain plasmid transmission and inter-plasmid integron transmission all playing important roles.
In addition to pathogen factors, we were able to analyse patient factors. While genomic surveillance previously focused on lineage-level analysis, long-read sequencing technologies have improved analysis of plasmids and other mobile genetic elements12,28,34. In our study, these insights proved informative both for understanding within-patient and between-patient blaIMP-4 spread. Within patients, we detected different trajectories of colonisation. Patients who undergo multiple colonisation events may be at the core of multiple transmission networks, as demonstrated by the patient colonised with two IncHI2A plasmids and highest betweenness centrality. Patients with inter-strain plasmid transfer may facilitate the diversification of genetic settings for blaIMP-4, thus increasing the risk of newly successful propagators emerging13, in turn fuelling outbreaks. Of note, we did not find clear evidence of blaIMP-4 integron transfer events within-patients, suggesting that they may play a lesser role. Long-read technologies also allowed us to analyse putative plasmid transmission between-patients, which implicated an additional 50% of wards, 44% transmission networks and 31% patients over strain transmission alone. We used detection of blaIMP-4 plasmids of the same MOB-typer primary/secondary clusters as a simple definition17 but quantitative thresholds incorporating changes in plasmid backbones and large-scale recombination events across a diverse array of bacterial hosts, plasmids and resistance determinants are needed35.
Our study had several limitations. Firstly, it was based on an isolate collection that spanned two decades with some patient data from the early part of the study being incomplete. Similarly, approaches to isolate sampling changed during that time, in particular since the inception of a statewide CPO detection programme that mandated screening in high-risk areas36 and likely led to increased detection of CPO colonisation in the last 4 years of the study. Finally, our study focused on blaIMP-4 and was from a single centre, which may limit the generalisability of findings to outbreaks at other centres.
In summary, we showed that blaIMP-4 endemicity and repeated outbreaks were due to diversification of genetic settings through inter-strain blaIMP-4 plasmid transfer and inter-plasmid blaIMP-4 integron transfer in combination with clonal expansion that led to an evolving cascade of high-risk strain-genetic setting pairs. Our findings provide a framework for understanding endemicity of MBL-producing organisms and may have broader applicability to other CPOs. Our study highlights that stopping the spread of CPOs will require adequate surveillance to detect not only the presence of resistance determinants and their bacterial host strains but also their genetic context and plasmid-integron transmission dynamics, thus enabling early detection of novel and potentially hidden threats.
Methods
The study was approved by the Alfred Hospital Ethics Committee (Project No: 44/20) with a waiver of consent for patient data due to its retrospective and observational nature.
Isolate selection
We systematically reviewed an institutional collection spanning all CPO isolates from 2002 to 2020. The collection contained isolates collected as part of routine clinical care, as well as environmental screening of sinks from 2018–2020. Routine antimicrobial susceptibility testing was performed using Vitek2 (BioMérieux). We identified blaIMP-4 carriage through polymerase chain reaction (PCR) screening. GoTaq Flexi DNA polymerase (Promega, Wisconsin, USA) was used as per manufacturer’s instructions and 10 µmol of the primers Imp4_screen_F (5′-CCAGGACACACTCCAGATAACC-3′) and Imp4_screen_R (5′-CAAGAGTGATGCGTCTCCAGC-3′) in 25 μL reaction volumes. PCR was performed using the following cycle conditions: 98 °C for 2 min, followed by 30 cycles of 98 °C for 30 sec, 55 °C for 30 sec, 72 °C for 30 sec. Amplicons were resolved by agarose gel electrophoresis on a 1% w/v agarose gel.
We selected 277 blaIMP-4 isolates for whole genome sequencing (WGS) based on bacterial strain (species/MLST combination) and year of isolation. For species with <30 isolates, we sequenced all available isolates. For species with >30 isolates, we performed WGS on selected isolates based on collection date to ensure that we had sequencing data available for all study periods. We sequenced at least one isolate of all strains across all study periods with both short-read (Illumina) and long-read (Oxford Nanopore) technologies (n = 172). We also selected one carbapenem-susceptible E. hormaechei ST93 and one E. hormaechei ST190 isolate for short- and long-read WGS.
Culture, DNA extraction and sequencing
All bacterial isolates were grown on cation-adjusted Mueller-Hinton II agar (Becton-Dickinson) for 16 h at 37 °C, and sub-cultured into cation-adjusted Mueller-Hinton broth (Becton-Dickinson) for a further 16 h at 37 °C. Bacterial genomic DNA was extracted from liquid culture using the GenFind V3 Reagent Kit (Beckman Coulter) as per manufacturer’s instructions. Libraries for short read sequencing were prepared using the Nextera Flex DNA Library Prep Kit (Illumina), and 150 bp paired-end sequencing was performed on the NovaSeq 6000 system (Illumina). Libraries for long-read sequencing were prepared using the Ligation Sequencing Kit with Native Barcoding Expansion (Oxford Nanopore Technologies) and sequenced on the MinION instrument with an R9.4.1 flow cell (Oxford Nanopore Technologies) for 48 h. Basecalling was performed with Guppy v.4.0.14 using the ‘high accuracy’ basecalling model.
De novo assembly and annotation
We constructed de novo assemblies of all isolates with only short-read data using the Shovill v1.0.4 wrapper for SPAdes, which also utilizes Trimmomatic for read trimming and Pilon for read error correction37,38,39,40. For long-read assembly, long reads were filtered using Filtlong v.0.2.041 with the following parameters: ‘--min_length 1000 --keep_percent 90 --target_bases 500000000’. Hybrid assemblies incorporating short- and long-read data were created using Unicycler v.0.4.08 with standard parameters42 with Unicycler output used to assess circularisation. If blaIMP-4 contigs were non-circularised, we re-assembled genomes using a long-read-first assembly using a bespoke pipeline (https://github.com/HughCottingham/clinopore-nf) that incorporates Flye v2.9.2 with subsequent polishing with Medaka v1.8.0, Polypolish v0.5.0 and Polca v3.4.143,44,45,46. Assembly quality was checked using Quast47 v5.2.0 and species identification was performed using GTDB-Tk48 v1.0.2 and checked against isolate identification performed at time of isolate collection. We excluded genomes (n = 7) with a species mismatch, as well as genomes whose assemblies had >1000 contigs, N50 < 10,000 or assembly length >7.5 Mb.
On the remaining assemblies (n = 270), we annotated the genomes using Prokka v1.14.649. We then performed resistance gene and plasmid replicon detection with Abricate v.1.0.050, using the NCBI Antibiotic Resistance and PlasmidFinder databases, respectively. We determined in silico multi-locus sequence type (ST) using ‘mlst’ v.2.19.051. All inconclusive ST calls with ‘mlst’ were checked with SRST252 v0.2.0.
Core genome-based phylogenetic analyses
We performed core genome-based phylogenetic analyses on key STs, defined as those with ≥5 isolates available from our institution. This included E. hormaechei ST93, ST114, and ST190 and P. aeruginosa ST111. Due to the absence of an MLST schema for S. marcescens we identified all RefSeq S. marcescens genomes and used Assembly Dereplicator v0.1.0 (https://github.com/rrwick/Assembly-Dereplicator) with a Mash distance threshold of 0.001 to remove duplicate assemblies. We then used these assemblies, in conjunction with S. marcescens genomes from our institution to construct a phylogeny using Mashtree18 v1.2.0. In brief, this tool uses non-alignment based assessment of sequence similarity through use of the min-hash algorithm, as implemented in Mash53, to generate distance metrics between input sequences. These are then used to cluster sequences using the neighbour joining algorithm. This allowed us to identify that Alfred Hospital genomes belonged to two lineages, for which we conducted the same phylogenetic analyses as we did within STs for other species.
This consisted of identifying RefSeq genomes of the same ST and including them for context in phylogenetic analyses. We chose one completed, closed assembly from our institution for each ST to use as a reference. Mobile genetic elements were excluded from these reference assemblies using PHASTER and IslandViewer 454,55. A core chromosomal SNV alignment was generated using Snippy v.4.6.056 and recombination was removed using Gubbins57 v3.3. We then used this core genome alignment in IQtree v.2.0.3 to generate maximum likelihood phylogenies for each ST58, with the best-fit model chosen using ModelFinder59. For each ST, median SNV distances between isolates from our institution were then calculated. Phylogenetic trees were visualized and annotated with metadata using ‘ggtree’60 with additional editing in Adobe Illustrator v2020.24.3.
Plasmid phylogenetic analyses
Using Abricate, we identified blaIMP-4-harbouring contigs that were putative plasmids in our hybrid assemblies. We then used the MOB-typer v1.4.9 tool to determine plasmid replicons present, as well as to assign clusters17. In addition, we used COPLA61 to assign plasmid taxonomic units to key plasmid types as determined by MOB-typer. We identified possible mosaic plasmids resulting from fusion events by examining plasmid replicon content within MOB-typer cluster and identifying plasmids which had presence of additional plasmid replicons then manually inspecting the assemblies.
We then conducted analyses within key plasmid groups within our dataset, as determined by MOB-typer cluster. These included IncC, IncHI2A type 1, IncHI2A type 2, IncFIA/IncFIB/IncP, IncFIB, IncL/M, and untypeable plasmids from Acinetobacter spp. In order to identify SNVs in the plasmid backbone, we used Snippy v.4.6.056 to create a core SNV alignment by mapping short reads to a reference plasmid from our institution from each plasmid group. We then used Mashtree18 to generate distance metrics between plasmids belonging to the same group, excluding mosaic plasmids. The R package ‘ggtree’ v3.0.4 was used to visualize the resulting trees60 and to annotate with metadata. Adobe Illustrator v2020.24.3 was used to merge different parts of the figures together. We also used fastANI v1.3 to generate pairwise average nucleotide identities between plasmids belonging to the same plasmid group62. We used progressiveMauve v2.4.0.r4736 to align all plasmids within a plasmid group and assess for structural re-arrangements63, then visualized this in Easyfig v2.2.264.
Analysis of bla IMP-4 integron and flanking sequences
We used Flanker19 v0.1.5 to identify and cluster flanking sequences around blaIMP-4 from hybrid contigs. We performed clustering 5000 bp upstream and downstream of the blaIMP-4 gene across windows in 500 bp increments. Geneious v10.2.6 (https://www.geneious.com) was used to visualize and assess for structural re-arrangements, with subsequent manual annotation in Adobe Illustrator v2020.24.3. We also assessed for SNVs in the blaIMP-4 integron by aligning blaIMP-4 genetic settings from completed, circularised assemblies to a previously reported blaIMP-4 integron (GenBank accession number JX101693)4 using MUSCLE v3.8.155165. Assemblies with large scale insertions or deletions in the integron were excluded (e.g. P. aeruginosa ST111 genomes). We extracted SNVs from the resulting alignment using SNP-sites v2.5.166 and grouped plasmids according to the SNV profile.
Short-read mapping to plasmid sequences
For genomes which only had short-read data available, we created a database of plasmids from all MOB-typer clusters (described above) and used the Nextflow implementation of the REDDog pipeline (V1.beta10.3; available at https://github.com/scwatts/reddog-nf) to map short-reads to this database. We used the following parameters: ‘mapping_cover_min = 1, mapping_mapped_min = 0.5, mapping_depth_min = 10’ then analysed the data. A read set was considered to have a match to a plasmid in the database if there was >90% coverage of the plasmid with <10 SNVs.
Patient data and transmission events
Clinical data were extracted from the electronic medical record. Clinical data were missing for 7 isolates from 2009–2012. Patient movement data were available from 2013 onwards, including 127/196 (65%) patients in the study. As patients did not undergo systematic surveillance for blaIMP-4, we considered that the patient may have been colonised in the 30 days prior to the first isolation of a blaIMP-4-harbouring organism and identified overlaps on the same ward at the same time as potential transmission events between patients. We then applied genomic criteria to further confirm potential transmission events. In the first instance, patients would have to have blaIMP-4-harbouring bacteria of the same strain for a potential transmission event to be considered. These events were then further classified on basis of SNV distance, with a cutoff of 10 SNVs. In the second instance, patients would have to have blaIMP-4 in the same genetic setting (defined as the same MOB-typer primary/secondary cluster), as determined either through completed assemblies or by having a match to a reference plasmid using the short-read mapping approach described above. We then used the R package ‘ggraph’ v2.0.5 to visualize putative transmission networks with patients as nodes and potential transmission events as edges. Betweenness centrality was calculated using the ‘betweenness’ function in the ‘iGraph’ R package (v1.2.11)67.
Statistical analysis
Categorical variables were compared using χ2 or Fisher’s exact tests and continuous variables were compared using Student’s t-test or Mann–Whitney–Wilcoxon, as appropriate. Statistical analyses were performed in R (v4.1.1).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The Illumina/Nanopore read data generated in this study have been deposited in the NCBI Sequence Read Archive under project accession PRJNA924056. The completed genome assemblies are available in GenBank; accessions are listed in Supplementary Dataset 1. The additional phylogenetic data generated in this study are provided in the Source Data file. Source data are provided with this paper.
Code availability
The code generated during this study is available on GitHub (https://github.com/nenadmacesic/imp4_ncomms)68.
References
Espedido, B. A., Partridge, S. R. & Iredell, J. R. bla(IMP-4) in different genetic contexts in Enterobacteriaceae isolates from Australia. Antimicrob. Agents Chemother. 52, 2984–2987 (2008).
Kizny Gordon, A. et al. Genomic dynamics of species and mobile genetic elements in a prolonged blaIMP-4-associated carbapenemase outbreak in an Australian hospital. J. Antimicrob. Chemother. 75, 873–882 (2020).
Logan, L. K. & Weinstein, R. A. The epidemiology of carbapenem-resistant enterobacteriaceae: the impact and evolution of a global menace. J. Infect. Dis. 215, S28–S36 (2017).
Partridge, S. R., Ginn, A. N., Paulsen, I. T. & Iredell, J. R. pEl1573 carrying blaIMP-4, from Sydney, Australia, is closely related to other IncL/M plasmids. Antimicrob. Agents Chemother. 56, 6029–6032 (2012).
Peleg, A. Y., Franklin, C., Bell, J. & Spelman, D. W. Emergence of IMP-4 metallo-beta-lactamase in a clinical isolate from Australia. J. Antimicrob. Chemother. 54, 699–700 (2004).
Peleg, A. Y., Franklin, C., Bell, J. M. & Spelman, D. W. Dissemination of the metallo-beta-lactamase gene blaIMP-4 among gram-negative pathogens in a clinical setting in Australia. Clin. Infect. Dis. 41, 1549–1556 (2005).
Peleg, A. Y., Franklin, C., Walters, L. J., Bell, J. M. & Spelman, D. W. OXA-58 and IMP-4 carbapenem-hydrolyzing beta-lactamases in an Acinetobacter junii blood culture isolate from Australia. Antimicrob. Agents Chemother. 50, 399–400 (2006).
Roberts, L. W. et al. Genomic analysis of carbapenemase-producing Enterobacteriaceae in Queensland reveals widespread transmission of bla(IMP-4) on an IncHI2 plasmid. Micro. Genom. 6, e000321 (2020).
Roberts, L. W. et al. Integrating multiple genomic technologies to investigate an outbreak of carbapenemase-producing Enterobacter hormaechei. Nat. Commun. 11, 466 (2020).
Hendrickx, A. P. A. et al. bla(OXA-48)-like genome architecture among carbapenemase-producing Escherichia coli and Klebsiella pneumoniae in the Netherlands. Micro. Genom. 7, 000512 (2021).
Marsh, J. W. et al. Evolution of outbreak-causing carbapenem-resistant Klebsiella pneumoniae ST258 at a Tertiary Care Hospital over 8 years. Mbio 10, e01945–19 (2019).
David, S. et al. Integrated chromosomal and plasmid sequence analyses reveal diverse modes of carbapenemase gene spread among Klebsiella pneumoniae. Proc. Natl Acad. Sci. USA 117, 25043–25054 (2020).
Leon-Sampedro, R. et al. Pervasive transmission of a carbapenem resistance plasmid in the gut microbiota of hospitalized patients. Nat. Microbiol. 6, 606–616 (2021).
Macesic, N. et al. Silent spread of mobile colistin resistance gene mcr-9.1 on IncHI2 ‘superplasmids’ in clinical carbapenem-resistant Enterobacterales. Clin. Microbiol. Infect. 27, 1856 e1857–1856 e1813 (2021).
Herbert, S. et al. Large outbreak of infection and colonization with gram-negative pathogens carrying the metallo- beta -lactamase gene blaIMP-4 at a 320-bed tertiary hospital in Australia. Infect. Control Hosp. Epidemiol. 28, 98–101 (2007).
Partridge, S. R., Kwong, S. M., Firth, N. & Jensen, S. O. Mobile genetic elements associated with antimicrobial resistance. Clin. Microbiol. Rev. 31, 1079–1061 (2018).
Robertson, J., Bessonov, K., Schonfeld, J. & Nash, J. H. E. Universal whole-sequence-based plasmid typing and its utility to prediction of host range and epidemiological surveillance. Micro. Genom. 6, mgen000435 (2020).
Katz, L. S. et al. Mashtree: a rapid comparison of whole genome sequence files. J. Open Source Softw. 4, 1762–1766 (2019).
Matlock, W. et al. Flanker: a tool for comparative genomics of gene flanking regions. Micro. Genom. 7, 000634 (2021).
Yoon, E. J. & Jeong, S. H. Mobile carbapenemase genes in Pseudomonas aeruginosa. Front. Microbiol. 12, 614058 (2021).
Annavajhala, M. K., Gomez-Simmonds, A. & Uhlemann, A. C. Multidrug-resistant Enterobacter cloacae complex emerging as a global, diversifying threat. Front. Microbiol. 10, 44 (2019).
Freeman, L. C. A set of measures of centrality based on betweenness. Sociometry 40, 35–41 (1977).
Nordmann, P. & Poirel, L. Epidemiology and diagnostics of carbapenem resistance in Gram-negative Bacteria. Clin. Infect. Dis. 69, S521–S528 (2019).
Cerqueira, G. C. et al. Multi-institute analysis of carbapenem resistance reveals remarkable diversity, unexplained mechanisms, and limited clonal outbreaks. Proc. Natl Acad. Sci. USA 114, 1135–1140 (2017).
Stoesser, N. et al. Genomic epidemiology of complex, multispecies, Plasmid-Borne bla(KPC) carbapenemase in enterobacterales in the United Kingdom from 2009 to 2014. Antimicrob. Agents Chemother. 64, e02244–19 (2020).
Babiker, A. et al. Clinical and genomic epidemiology of carbapenem-nonsusceptible citrobacter spp. at a Tertiary Health Care center over 2 decades. J. Clin. Microbiol 58, e00275–20 (2020).
Dolejska, M., Papagiannitsis, C. C., Medvecky, M., Davidova-Gerzova, L. & Valcek, A. Characterization of the complete nucleotide sequences of IMP-4-encoding plasmids, belonging to diverse Inc families, recovered from enterobacteriaceae isolates of wildlife origin. Antimicrob. Agents Chemother. 62, e02434–02417 (2018).
Boonyasiri, A. et al. Integrated patient network and genomic plasmid analysis reveal a regional, multi-species outbreak of carbapenemase-producing Enterobacterales carrying both bla-IMP and mcr-9 genes. Medrxiv https://doi.org/10.1101/2021.10.28.21265436 (2021).
Constantinides, B. et al. Genomic surveillance of Escherichia coli and Klebsiella spp. in hospital sink drains and patients. Microb. Genom. https://doi.org/10.1099/mgen.0.000391 (2020).
Snitkin, E. S. Contamination of Hospital plumbing: a source or a sink for antibiotic-resistant organisms? Jama Netw. Open 2, e187660 (2019).
Stoesser, N. et al. Genomic epidemiology and longitudinal sampling of ward wastewater environments and patients reveals complexity of the transmission dynamics of blaKPC-carbapenemase-producing Enterobacterales in a hospital setting. Medrxiv https://doi.org/10.1101/2021.11.26.21266267 (2021).
Weingarten, R. A. et al. Genomic analysis of hospital plumbing reveals diverse reservoir of bacterial plasmids conferring carbapenem resistance. Mbio 9, e02011-02017 - 02016 (2018).
Kotsanas, D. et al. “Down the drain”: carbapenem‐resistant bacteria in intensive care unit patients and handwashing sinks. Med. J. Aust. 198, 267–269 (2013).
Evans, D. R. et al. Systematic detection of horizontal gene transfer across genera among multidrug-resistant bacteria in a single hospital. Elife 9, e53886 (2020).
Hawkey, J. et al. ESBL plasmids in Klebsiella pneumoniae: diversity, transmission, and contribution to infection burden in the hospital setting. Medrxiv https://doi.org/10.1101/2021.12.20.21268000 (2021).
Lane, C. R. et al. Search and contain: impact of an integrated genomic and epidemiological surveillance and response program for control of carbapenemase-producing enterobacterales. Clin. Infect. Dis. 73, e3912–e3920 (2021).
Seemann, T. Shovill: Faster SPAdes (or better SKESA/Megahit/Velvet) Assembly of Illumina Reads. https://github.com/tseemann/shovill (2018).
Bankevich, A. et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477 (2012).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Walker, B. J. et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One 9, e112963 (2014).
Wick, R. R. Filtlong. https://github.com/rrwick/Filtlong (2020).
Wick, R. R., Judd, L. M., Gorrie, C. L. & Holt, K. E. Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput Biol. 13, e1005595 (2017).
Kolmogorov, M., Yuan, J., Lin, Y. & Pevzner, P. A. Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 37, 540–546 (2019).
Wick, R. R. & Holt, K. E. Polypolish: Short-read polishing of long-read bacterial genome assemblies. PLoS Comput. Biol. 18, e1009802 (2022).
Wick, R. R., Judd, L. M. & Holt, K. E. Assembling the perfect bacterial genome using Oxford Nanopore and Illumina sequencing. PLoS Comput. Biol. 19, e1010905 (2023).
Zimin, A. V. & Salzberg, S. L. The genome polishing tool POLCA makes fast and accurate corrections in genome assemblies. PLoS Comput. Biol. 16, e1007981 (2020).
Gurevich, A., Saveliev, V., Vyahhi, N. & Tesler, G. QUAST: quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075 (2013).
Chaumeil, P. A., Mussig, A. J., Hugenholtz, P. & Parks, D. H. GTDB-Tk: a toolkit to classify genomes with the Genome Taxonomy Database. Bioinformatics 36, 1925–1927 (2019).
Seemann, T. Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069 (2014).
Abricate: mass screening of contigs for antimicrobial resistance or virulence genes (2020).
Seemann, T. mlst: Scan contig files against traditional PubMLST typing schemes. (2020).
Inouye, M. et al. SRST2: Rapid genomic surveillance for public health and hospital microbiology labs. Genome Med. 6, 90 (2014).
Ondov, B. D. et al. Mash: fast genome and metagenome distance estimation using MinHash. Genome Biol. 17, 132 (2016).
Arndt, D. et al. PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res. 44, W16–W21 (2016).
Bertelli, C. et al. IslandViewer 4: expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res. 45, W30–W35 (2017).
Seemann, T. Snippy: Rapid Haploid Variant Calling and Core SNP Phylogeny. https://github.com/tseemann/snippy (2020).
Croucher, N. J. et al. Rapid phylogenetic analysis of large samples of recombinant bacterial whole genome sequences using Gubbins. Nucleic Acids Res. 43, e15 (2015).
Minh, B. Q. et al. IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 37, 1530–1534 (2020).
Kalyaanamoorthy, S., Minh, B. Q., Wong, T. K. F., von Haeseler, A. & Jermiin, L. S. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat. Methods 14, 587–589 (2017).
Yu, G. C., Smith, D. K., Zhu, H. C., Guan, Y. & Lam, T. T. Y. GGTREE: an R package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol. Evol. 8, 28–36 (2017).
Redondo-Salvo, S. et al. COPLA, a taxonomic classifier of plasmids. BMC Bioinforma. 22, 390 (2021).
Jain, C., Rodriguez, R. L., Phillippy, A. M., Konstantinidis, K. T. & Aluru, S. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 9, 5114 (2018).
Darling, A. E., Mau, B. & Perna, N. T. progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One 5, e11147 (2010).
Sullivan, M. J., Petty, N. K. & Beatson, S. A. Easyfig: a genome comparison visualizer. Bioinformatics 27, 1009–1010 (2011).
Edgar, R. C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 (2004).
Page, A. J. et al. SNP-sites: rapid efficient extraction of SNPs from multi-FASTA alignments. Micro. Genom. 2, e000056 (2016).
Csardi, G. & Nepusz, T. The igraph software package for complex network research. Int. J. Complex Syst. 1695, 1–9 (2006).
Macesic, N. et al. Genomic dissection of endemic carbapenem resistance reveals metallo-beta-lactamase dissemination pathways through clonal, plasmid and integron transfer. Zenodo https://doi.org/10.5281/zenodo.8051453 (2023).
Acknowledgements
This work was supported by the National Health and Medical Research Council of Australia (Emerging Leader 1 Fellowship APP1176324 to N.M. and Practitioner Fellowship APP1117940 to A.Y.P.). The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Author information
Authors and Affiliations
Contributions
N.M. and A.Y.P. conceived the study. J.A.W., A.D., D.W.S. and A.W.J.J. designed and supervised sampling and collection of bacterial isolates. L.V.B., T.H., K.P. and G.Z.B. collected the bacterial isolates, performed bacterial characterisation and conducted whole genome sequencing of isolates. N.M. collected all clinical data. N.M., J.H., B.V. and H.C. performed bioinformatics analyses. N.M., J.H. and A.Y.P. analysed all results. N.M. wrote the initial draft of the manuscript. N.M., J.H. and A.Y.P. contributed to the final version of the manuscript. All authors read and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
N.M. has received research support from GlaxoSmithKline, unrelated to the current study. A.Y.P. has received research funding from MSD through an investigator-initiated research project. All other authors declare no conflict of interest.
Additional information
Peer review information Nature Communications thanks Alvaro San Millan and the other anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Macesic, N., Hawkey, J., Vezina, B. et al. Genomic dissection of endemic carbapenem resistance reveals metallo-beta-lactamase dissemination through clonal, plasmid and integron transfer. Nat Commun 14, 4764 (2023). https://doi.org/10.1038/s41467-023-39915-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-023-39915-2
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
