
Abstract
Mutations in TUB-like protein 1 (TULP1) are associated with severe early-onset retinal degeneration in humans. However, the pathogenesis remains largely unknown. There are two homologous genes of TULP1 in zebrafish, namely tulp1a and tulp1b. Here, we generated the single knockout (tulp1a−/− and tulp1b−/−) and double knockout (tulp1-dKO) models in zebrafish. Knockout of tulp1a resulted in the mislocalization of UV cone opsins and the degeneration of UV cones specifically, while knockout of tulp1b resulted in mislocalization of rod opsins and rod-cone degeneration. In the tulp1-dKO zebrafish, mislocalization of opsins was present in all types of photoreceptors, and severe degeneration was observed at a very early age, mimicking the clinical manifestations of TULP1 patients. Photoreceptor cilium length was significantly reduced in the tulp1-dKO retinas. RNA-seq analysis showed that the expression of tektin2 (tekt2), a ciliary and flagellar microtubule structural component, was downregulated in the tulp1-dKO zebrafish. Dual-luciferase reporter assay suggested that Tulp1a and Tulp1b transcriptionally activate the promoter of tekt2. In addition, ferroptosis might be activated in the tulp1-dKO zebrafish, as suggested by the up-regulation of genes related to the ferroptosis pathway, the shrinkage of mitochondria, reduction or disappearance of mitochondria cristae, and the iron and lipid droplet deposition in the retina of tulp1-dKO zebrafish. In conclusion, our study establishes an appropriate zebrafish model for TULP1-associated retinal degeneration and proposes that loss of TULP1 causes defects in cilia structure and opsin trafficking through the downregulation of tekt2, which further increases the death of photoreceptors via ferroptosis. These findings offer insight into the pathogenesis and clinical treatment of early-onset retinal degeneration.
Similar content being viewed by others


Introduction
TULP1, also known as Leber congenital amaurosis 15 (LCA15) or retinitis pigmentosa-14 (RP14) [1], has been associated with autosomal recessive early-onset retinal degeneration [2,3,4,5,6,7,8]. TULP1 was first reported as a pathogenic gene of retinal degeneration in 1998 [2, 3]. At the present time, 99 pathogenic mutations have been reported (http://www.hgmd.cf.ac.uk/ac/gene.php?gene=TULP1). Most of the affected individuals display congenital nystagmus, night blindness, and severely reduced visual acuity in their first year of life [4, 6, 9]. Revealing the function of TULP1 is crucial for understanding the pathogenesis of TULP1-associated retinal degeneration.
TULP1 is a member of the tubby family proteins that contain a highly conserved tubby domain at the carboxyl-terminal. This protein family includes TUB, TULP1, TULP2, and TULP3 [10, 11]. Through a structure-directed approach, Boggon et al. predicted that Tubby-like proteins are a unique family of bipartite transcription factors [12]. Whether tubby proteins can regulate transcription under physiological conditions remains to be confirmed. In COS7 cells, TULP1 is localized to the plasma membrane and nucleus [13]. Another tubby family protein, tubby, is found primarily in the nucleus in a primary culture of hippocampal neurons [12]. In the retina, TULP1 is a photoreceptor-specific protein localized to the inner segments, connecting cilium, and synaptic terminals [14, 15]. Deletion of Tulp1 in mice causes severe retinal degeneration and deficient protein transport in the photoreceptors [15,16,17]. The exact mechanism by which TULP1 regulates protein trafficking through the cilium is not well understood.
The photoreceptor cilium is supported by a microtubule-based axoneme backbone. Defects in the structure and/or function of photoreceptor cilium lead to a broad range of retinal dystrophy [18,19,20]. The involvement of TULP3 in ciliary protein transport has been extensively studied. TULP3 can directly bind GPCRs and facilitate their trafficking to the cilia [21]. TULP3 is also critical for cilia formation in hRPE-1 cells [22]. As for TULP1, a previous study demonstrated that the specific ciliary localization of photoreceptor disc component (PRCD), a protein involved in the formation of photoreceptor OS discs, is dependent on TULP1 and confirmed the interaction between PRCD and TULP1 [23]. Currently, the function of TULP1 and the exact etiology of how TULP1 mutations cause early-onset retinal degeneration have not been clearly established.
Ferroptosis is characterized by the iron-dependent accumulation of lipid peroxidation [24]. In recent years, there has been increasing evidence that ferroptosis also plays a role in neurodegenerative diseases [25]. Treatment with iron chelators could increase the survival of photoreceptors in light-induced photoreceptor degeneration and retinitis pigmentosa mouse models [26, 27]. However, the possible involvement of ferroptosis in the death of photoreceptors in inherited retinal degeneration has not been fully studied.
In recent years, due to a number of benefits, zebrafish has become a popular animal model by which to study the pathogenic mechanism of retinal diseases [28]. In particular, in vitro fertilization and embryonic development of zebrafish allows real-time observation of organogenesis occurring at very early embryonic stages. Given the severe visual phenotype associated with TULP1 patients from infancy, it is necessary to study the retinal changes before and after lesions. In this work, we generated a zebrafish tulp1 knock-out model and found that Tulp1a and Tulp1b play an important role in the assembly of cilium in photoreceptors. Tulp1a and Tulp1b could increase the transcription activity of the tekt2 promoter in ZF4 cells, suggesting that TULP1 functions as a transcription factor. In addition, our study highlights ferroptosis as a novel degeneration manner in early-onset retinal degeneration.
Results
Generation of the tulp1a and tulp1b knockout zebrafish line via CRISPR-Cas9 technique
Sequence alignment showed that there are two orthologous genes of TULP1 in zebrafish, namely tulp1a and tulp1b. Zebrafish Tulp1a and Tulp1b show, respectively, about 72.45% and 70.94% alignment in the amino acid residues with human TULP1 (Fig. S1A). The predicted 3D structures of tubby domain show structural similarities between zebrafish and human (Fig. S1B). Our previous single-cell sequencing data on zebrafish retinas [29] showed that tulp1a and tulp1b are expressed in all types of photoreceptors, while tulp1a shows a higher expression than tulp1b in cones, particularly in the UV/blue cones (Fig. S2, Table S4).
The pathogenic mutations found in TULP1 were distributed in all exons except for exon 9 (Fig. S1C). To explore the function of TULP1, we used CRISPR-Cas9 to generate knockout lines of tulp1a and tulp1b. The target sites aligned with the exon 4 and exon 8 of TULP1 in human, respectively (Fig. 1A, Fig. S1C). After three generations’ screening, we chose one tulp1a mutant line (tulp1a−/−) (c.115_118delGGTG, p.Gly39Metfs*12) and one tulp1b mutant line (tulp1b−/−) (c.591delC, p.Gly198Glufs*27) for subsequent study (Fig. 1B). A tulp1a and tulp1b double knockout zebrafish line (tulp1-dKO) was generated by crossing the two mutant lines. The mRNA levels of tulp1a and tulp1b were markedly reduced in, respectively, tulp1a−/−, tulp1b−/−, and in tulp1-dKO zebrafish (Fig. 1C). The temporal and spatial expression patterns of tulp1a and tulp1b were further determined by whole-mount in situ hybridization (WISH). At 24–48 hour post-fertilization (hpf), both genes were specifically expressed in the pineal gland. Subsequent to 48 hpf, tulp1a and tulp1b were observed to be expressed in the retina, whereas in tulp1-dKO zebrafish the expression of tulp1a and tulp1b was significantly decreased from 48 hpf onward (Fig. 1D, E). The Tulp1 protein could not be detected in tulp1-dKO zebrafish (Fig. 1F). Collectively, these results suggest our knockout zebrafish lines are successful.

A The gene structure and CRISPR/Cas9 target sites are shown. The numbers represent exons. B DNA sequencing showing the tulp1a mutant line (tulp1a−/−) (c.115_118delGGTG), and tulp1b mutant line (tulp1b−/−) (c.591delC). C Decreased expression of tulp1a or tulp1b was detected by qRT-PCR in, respectively, tulp1a−/−, tulp1b−/−, and in tulp1-dKO zebrafish at 4 dpf. 18s-rRNA was used as an endogenous control (n = 3). Mean ± SD. **P < 0.01. ****P < 0.0001. D, E Whole-mount in situ hybridization showing the expression pattern of tulp1a and tulp1b in wt and tulp1-dKO zebrafish. (n = 20). Scale bar: 50 µm. F The protein level of Tulp1 was undetectable at 4 mpf (n = 3). dpf: day post-fertilization; mpf: month post-fertilization.
Photoreceptor degeneration in the tulp1a and tulp1b knockout zebrafish
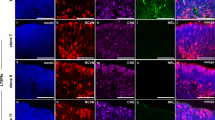
Hematoxylin and Eosin staining results showed that no significant difference was observed in retina between wild-type (wt) and tulp1a−/− zebrafish (Fig. S3A). To explore possible differences in photoreceptor types, we labeled rods and cones (red, green, blue, and UV cones) with specific antibodies (Rhodopsin, Opn1lw1, Opn1mw1, Opn1sw2, and Opn1sw1) in retinal sections. There are no differences in any of the other photoreceptor types except UV cones. In tulp1a−/− zebrafish, at 3 day post-fertilization (dpf), the outer segments (OSs) of UV cones were shorter than those of wt zebrafish and were almost undetectable as early as 7 dpf (Fig. S3B and C). Western blot also showed no obvious difference in the expression of rod marker-Gnat1, even at 24 month post-fertilization (mpf), whereas the expression of cone marker-Gnat2 was slightly downregulated (Fig. S3D and E). These results indicate that only UV cones were degenerated in tulp1a−/− zebrafish.
Compared to tulp1a−/− mutants, significant photoreceptor degeneration was detected in tulp1b−/− zebrafish, with the degree of degradation increasing with age. The outer nuclear layer (ONL) of tulp1b−/− zebrafish was thinner and the OSs were shorter compared to those of wt zebrafish (Fig. S4A and B). Immunofluorescence results showed that in the tulp1b−/− zebrafish, the length of rods OSs was obviously shorter as early as 3 dpf (Fig. S4C). Western blot showed that in tulp1b−/− mutants the expression of Gnat1 was barely detectable at 1 mpf, while the downregulation of Gnat2 was not as significant as that of Gnat1 (Fig. S4D and E). These results suggest that tulp1b knockout led to a rod-cone degeneration phenotype.
Next, we examined the phenotype of the retina in tulp1-dKO zebrafish at the indicated ages. The signals of photoreceptor markers were markedly reduced at each age. By 5 dpf, the photoreceptor layer was almost absent from the central retina in tulp1-dKO zebrafish (Fig. 2A). The fine structure of photoreceptors was examined at 5 dpf using transmission electron microscopy (TEM). In wt retinas, membrane discs in OSs were well-organized. However, in tulp1-dKO retinas, the disc membranes were disorganized and contained concentric circular structures (Fig. 2B). Consistent with the above results, the mRNA and protein levels of Gnat1 and Gnat2 were significantly decreased (Fig. 2C and D). These results suggest that the function of photoreceptors was impaired at a very early stage in tulp1-dKO zebrafish, which is similar to the clinical phenotype of early-onset retinal degeneration [30].

A Sections were stained with rod-specific marker Rhodopsin, green cone-specific marker Opn1mw1, red cone-specific marker Opn1lw1, blue cone-specific marker Opn1sw2, and UV cone-specific marker Opn1sw1 at 3 dpf, 5 dpf, 20 dpf, and 4 mpf. ONL, outer nuclear layer; INL, inner nuclear layer; GCL, ganglion cell layer. Scale bar: 15 µm. B TEM of the photoreceptors of wt and tulp1-dKO zebrafish at 5 dpf. The higher-magnification images within the rectangles (in a and b) are shown on the right (a’ and b’). Scale bar: 2 µm. C Relative mRNA expression of gnat1 and gnat2 in wt and tulp1-dKO zebrafish at 4 dpf. Mean ± SD (n = 3). ****P < 0.0001. D Gnat1 and Gnat2 were detected by western blot.
Ciliary defects precede photoreceptor degeneration in tulp1-dKO zebrafish
Given the mislocalization of Opn1sw1 in tulp1a−/− zebrafish (Figure S3C), Rhodopsin in tulp1b−/− zebrafish (Fig. S4B and C), and all opsins in tulp1-dKO zebrafish (Fig. 2A), it might be surmised that TULP1 has a function similar to that of TULP3 in mediating ciliary trafficking by interacting with diverse cargos [21]. Interestingly, no colocalization of Tulp1b and Rhodopsin was found in HEK293 cells (Fig. S5A). Moreover, Tulp1a and Tulp1b were localized in the nucleus in HEK293T cells, ARPE-19 cells, and ZF4 cells (Fig. S5B). These findings suggest that Tulp1a and Tulp1b may not affect the localization of Rhodopsin through direct interaction. Previous studies have demonstrated that TULP3 affected the formation of cilia [22]. To investigate the role of Tulp1 in cilia formation, ciliogenesis was analyzed in wt and tulp1-dKO zebrafish. The results showed that reduced ciliogenesis and shorter cilia were observed at 68 hpf, 3 dpf, and 4 dpf in tulp1-dKO zebrafish compared with wt (Fig. 3A, B), suggesting that ciliogenesis was affected in tulp1-dKO zebrafish.

A Sections of wt and tulp1-dKO were stained with Ac-Tub at 60 hpf, 68 hpf, 3 dpf, and 4 dpf. Scale bar: 2 µm (n = 6). B Quantification of cilium lengths in wt and tulp1-dKO zebrafish presented in A. Mean ± SD. (n ≥ 40). ****P < 0.0001. Ac-Tub: Acetylated-α-Tubulin, the axonemal marker.
Tulp1a and Tulp1b affect ciliogenesis through promotion of tekt2 expression
To investigate the mechanism underlying the cilium defect and photoreceptor degeneration resulting from tulp1 deletion, we performed RNA-seq of wt zebrafish and tulp1-dKO zebrafish. A total of 895 genes were expressed significantly differently between wt and tulp1-dKO zebrafish, of which 303 genes were significantly upregulated and 592 genes were significantly downregulated in the tulp1-dKO zebrafish (Fig. 4A, B). Interestingly, we found that only three genes related to cilium (tekt2, arl3l2, cep126) were significantly reduced in the RNA-seq data (Fig. 4C). Consistent with the RNA-seq data, a marked reduction in the expression of tekt2, arl3l2, and cep126 was observed in the tulp1-dKO zebrafish at 4 dpf. Notably, however, only the expression of tekt2 was significantly reduced as early as 48 hpf (Fig. 4D). Herein, we speculate that tekt2 might be the target gene of Tulp1a and Tulp1b. The luciferase reporter assay results showed that Tulp1a and Tulp1b significantly increased the transcription activity of tekt2 promoter in ZF4 cells. Co-expression of Tulp1a and Tulp1b further increased the activity of tekt2 promotor in comparison with the expression of Tulp1a and Tulp1b separately (Fig. 4E). TEKT2 is a member of TEKTINs, a family of elongated proteins that assemble into extended filaments [31]. WISH showed that tekt2 was indeed expressed in eyes (Fig. S6). The downregulation of tekt2 may explain the shortened cilia length in tulp1-dKO zebrafish. Collectively, these results indicate that tekt2 was downregulated in tulp1-dKO zebrafish and that Tulp1a and Tulp1b could promote the expression of tekt2 in ZF4 cells.

Heatmap (A) and Volcano plot (B) for differential gene expression between wt and tulp1-dKO zebrafish. C Volcano plot displaying the gene expression related to cilium for wt versus tulp1-dKO. Genes with |log2FC| ≥ 1.5 and P-value ≤ 0.05 are highlighted in green. D The mRNA levels that significant enrichment in (C) were detected by qRT-PCR (n = 3). E Reporter plasmid containing the promotor of tekt2 and pEGFP-C1, pEGFP-Tulp1a, pEGFP-Tulp1b were transfected into ZF4 cells (n = 3). Mean ± SD. ***P < 0.001.
Ferroptosis was activated in photoreceptor cells of tulp1-dKO zebrafish
As shown in Fig. 2A, there were severe degeneration of photoreceptors in tulp1-dKO zebrafish. To investigate the mechanism of cell death, we performed Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis based on the RNA-seq data. KEGG analysis revealed that the phototransduction pathway is significantly enriched, which is consistent with the retina degeneration phenotype in tulp1-dKO zebrafish. Interestingly, the Arachidonic acid metabolism and Linoleic acid metabolism pathway were significantly enriched (Fig. 5A), which were considered to be involved in the ferroptosis [24, 32]. The Gene Set Enrichment Analysis (GSEA) also showed a tight correlation between ferroptosis and the tulp1-dKO gene sets (Fig. 5B). The iron-containing enzyme lipoxygenase is the main promotor of ferroptosis through the production of lipid hydroperoxides [33]. The expression of genes involved in the iron homeostasis and lipid peroxidation was verified by qRT-PCR (Fig. 5C–H). Consistent with RNA-seq data, the expression of ptgs2b, alox5b.3, cyp2p9, cyp2p10—the in vivo biomarkers for ferroptosis that contributes to lipid peroxidation [33, 34]—was significantly increased in the tulp1-dKO zebrafish (Fig. 5C–F). Furthermore, accumulation of lipid, detected using BODIPY and Nile red staining, was observed in the tulp1-dKO retina (Fig. 5I, J). In addition, we also detected iron accumulation by Perls/DAB staining. The results showed that there was markedly increased iron signaling in the photoreceptors of tulp1-dKO zebrafish compared with wt zebrafish at 4 dpf (Fig. 5K). Consistent with this iron accumulation, the expression of fthl28 and cp was upregulated in the tulp1-dKO zebrafish (Fig. 5G, H), suggesting a disruption in iron homeostasis [35,36,37]. ATF3 contributed to ferroptosis via increasing H2O2 and iron [38], while the expression of atf3 was upregulated in the tulp1-dKO zebrafish, suggested the activation of ferroptosis in tulp1-dKO zebrafish (Fig. S7). Morphology of mitochondria is also one of the features of ferroptosis [32]. TEM results showed mitochondria exhibited changes characteristic of ferroptosis, including shrinkage and disappearance of cristae in the inner segments of tulp1-dKO zebrafish compared with wt zebrafish (Fig. 5L). However, the expression levels of Gpx4a and Gpx4b were unchanged as detected by qRT-PCR and western blot (Fig. S8). Based on the above, we conclude that ferroptosis participates in the photoreceptor degeneration seen in tulp1-dKO zebrafish.

A KEGG pathway enrichment analysis of the genes identified to be differentially expressed between wt and tulp1-dKO groups. The significantly enriched pathways are shown. B GSEA of ferroptosis-related genes. C–H Relative expression of genes involved in ferroptosis was detected by qRT-PCR at 4 dpf. Mean ± SD. *P < 0.05. **P < 0.01. Lipid droplets performed by Nile Red (I) or BODIPY (J) in wt and tulp1-dKO zebrafish at 4 dpf. Enlarged images of the boxed areas are shown in the lower right corner. Scale bar: 20 µm. K Perls/DAB staining in wt and tulp1-dKO zebrafish at 4 dpf. Enlarged images of the boxed areas are shown beneath. Scale bar: 25 µm. L TEM images of mitochondria from wt and tulp1-dKO zebrafish. M, mitochondria. OS, outer segment. Scale bar: 0.2 µm.
Discussion
Shortened cilium in photoreceptors impedes the transporting of opsins and leads to the mislocalization in tulp1-dKO zebrafish. RNA-seq analysis and subsequent experiments suggest that downregulation of tekt2 might be the cause of the shortened cilium and indicates an important role of ferroptosis in photoreceptor degeneration. This is the first time that the effect on cilia formation of TULP1 depletion has been elucidated. In addition, in vitro luciferase assay confirmed the possibility of TULP1 as a transcription factor (Fig. 6).

Depletion of Tulp1a and Tulp1b in zebrafish diminished the transcriptional activity of tekt2, which impaired the ciliogenesis of photoreceptors, and thus interfering the trafficking of opsins. Eventually, ferroptosis was activated in photoreceptors, and retina was degenerated.
Loss of TULP1 results in severe early-onset retinal degeneration. Although many recent studies have explored the function of TULP1, there are some problems that remain unresolved: (1) Tubby proteins were predicted to be the transcription factors [12, 39], but the direct evidence for their role as transcription factors has not been reported; (2) Protein mislocalization has been reported in Tulp1 knockout mice, although how TULP1 mutation causes protein mislocalization has not been studied; (3) The mechanism of photoreceptor degeneration in TULP1 mutant patients is not clear.
In the present study, we utilized the zebrafish to investigate the function of TULP1 and to explore the above questions. In tulp1a−/− zebrafish, we observed, specific mislocalization of opsins in UV cones that then suffered degeneration. In tulp1b−/− zebrafish, there was a retinitis pigmentosa phenotype. The different phenotypes in the two models may be due to the different expression levels of the two genes in different photoreceptor cells, a possibility that can be resolved through further study. Lital et al. showed that TULP1 is required for specific localization of PRCD by direct interaction [23]. In contrast, in the present study, no colocalization of Tulp1b and Rhodopsin was found, suggesting that Tulp1 does not mediate Rhodopsin transport by direct interaction. Cross-sectional views of cilium had the normal 9 + 0 arrangement of microtubule pairs in mice [15], suggesting that the basal body of cilium is normal. In our study, we demonstrated that tulp1a and tulp1b affected the length of cilium in photoreceptors and caused subsequent mislocalization of proteins. Recently, Hong et al. suggested that TULP1 transfection could not rescue cilia formation and length in TULP3 KO RPE1 cells [22], a finding that is not consistent with our results probably due to differential requirements of TULP1 for ciliogenesis in each cell type.
Tekt2 is a member of Tektins, originally identified as structural components of axonemal doublet microtubules in cilia and flagella [31]. Human TEKT2 has been suggested to be an indicator of sperm motility loss [40]. Rebecca et al. demonstrated that one member of this family, Tektin-1, is a positive regulator of cilium length [41]. Our analysis showed that tekt2 was downregulated prior to the emergence of shortened cilium, fueling our speculation that tekt2 has specific functions in ciliogenesis in the photoreceptor. As Tulp1a and Tulp1b indeed enhanced the transcriptional activity of the tekt2 promoter, we surmise that tekt2 is one of the downstream effectors of Tulp1a and Tulp1b in zebrafish. However, the direct interaction and other target genes require further investigation. These results also indicated that tekt2 might be a candidate gene of retinal disease.
Iron dyshomeostasis and lipid peroxidation are features of ferroptosis. In addition to being a cofactor of iron-containing lipoxygenase, iron can also generate free radicals directly through the Fenton reaction, leading to lipid peroxidation [42, 43]. In our study, iron accumulation, lipid droplets, and abnormal mitochondria in retina were detected in the tulp1-dKO zebrafish. The genes involved in lipid peroxide, such as ptgs2b, alox5b.3, cyp2p9, cyp2p10, were considerably upregulated. Together, these results indicate that ferroptosis contributes to the death of photoreceptors in tulp1-dKO zebrafish. However, what causes the iron accumulation is unclear. Based on our own and other studies, we speculate that the disturbed visual cycle and phototransduction cascade may cause the disorder of iron homeostasis, as the enzymes involved in these two processes such as RPE65, GC, GCAP5 are iron-dependent [44]. In addition, abnormal connection between RPE and photoreceptors cause disordered iron homeostasis, as that iron is recycled through phagocytosis [45]. As for lipid accumulation: with the rapid degradation of photoreceptors, a large number of outer segments (containing high levels of polyunsaturated fatty acids), which could not be phagocytized and digested by RPE cells in a timely fashion, accumulated in the photoreceptor layer, so resulting in lipid accumulation. Caberoy et al. suggested that Tulp1 could facilitate phagocytosis through MerTK [46], such that knockout Tulp1 may decrease the effect of RPE phagocytosis. Masek et al. revealed that loss of the Bardet-Biedl syndrome 1 (important for ciliary transport) in zebrafish locally disrupted lipid homeostasis [47]. Here, in our model, abnormal ciliary transport may lead to disturbed lipid homeostasis. GPX4 is a central inhibitor of ferroptosis, GPX4 degradation or inhibition can promote ferroptosis [48]. The reason why the expression of Gpx4 was unaffected in our zebrafish tulp1-dKO model remains unclear. Possibly, there may another GPX family protein or GPX4-independent systems responsible for the inhibition of ferroptosis in retinal cells during retinal degeneration. Further studies are needed to demonstrate this assumption in the future. Given that iron-mediated ferroptosis is involved in the death of photoreceptors in the tulp1-dKO zebrafish, it is plausible that an iron-chelating agent may be useful in treatment. Our current study also raises an intriguing question as to a possible role of ferroptosis in retinal diseases caused by other genes.
In conclusion, we revealed an essential role of TULP1 in ciliogenesis and survival of photoreceptors. Our results showed that Tulp1a and Tulp1b might act as transcription factors to regulate the expression of tekt2 to affect ciliogenesis. The tulp1-dKO zebrafish line may be extremely useful in providing further insights into the physiological function of TULP1 in ciliogenesis and the role of ferroptosis in inherited retinal degeneration, and therefore act as a basis for new strategies for the prevention and treatment of such diseases.
Materials and methods
Zebrafish maintenance and breeding
The zebrafish were kept in the recirculating water system (pH 6.6–7.4, 26–28.5 °C) with a daily cycle of 14 h of light and 10 h of dark. The zebrafish embryos were collected and placed in E3 medium and kept in a 28.5 °C incubator until hatched. To avoid bias affecting the selection of zebrafish, we randomized the embryos for all experiments, no blinding was used. There were no inclusion or exclusion criteria used in the selection of the animals. All studies were approved by the Ethics Committee of Huazhong University of Science and Technology.
Generation of zebrafish knockout lines
The CRISPR/Cas9 system was purchased from the China Zebrafish Resource Center (CZRC). CRISPR/Cas9 target sites were designed using the online tool CHOPCHOP (http://chopchop.cbu.uib.no/) [49]. The single-guide RNAs (sgRNAs) and Cas9 mRNA were synthesized using the in vitro transcription kits (Thermo Scientific and Ambion). Wild-type (wt) zebrafish embryos at the one-cell stage were co-injected with the Cas9 mRNA and sgRNAs. The detailed experimental methods have been described previously [50].
Hematoxylin and eosin (H&E) staining
The eyes of zebrafish were fixed with 4% PFA for 3 h at room temperature (RT), dehydrated with 30% sucrose overnight at 4 °C, and then embedded in OCT and sectioned as described in previous studies [51]. The sections were stained with the Hematoxylin and Eosin Staining Kit (Beyotime, Shanghai, China). Images were captured by optical microscope BX53.
Immunofluorescence
The frozen sections were dried at RT, soaked in PDT (PBS containing 1% DMSO and 0.1% TritonX-100) twice for ten minutes to remove the OCT, then blocked with PBDT (PDT containing 1%BSA) containing 10% goat serum for 1 h at RT. After removing the blocking solution, the slides were incubated with the primary antibody overnight at 4 °C (Nile Red and BODIPY staining were incubated for 10 min at RT). The slides were washed three times with PDT and incubated with the secondary antibody of the corresponding species for 1 h at 37 °C in the dark. Following by staining the cell nucleus with DAPI and washing with PBS, the sections were mounted with 50% glycerol in PBS. The fluorescence images were captured with a confocal laser-scanning microscope (FV3000, Olympus). The list of the primary antibodies is provided in Table S1.
Western blot
About 35 heads of zebrafish larvae age of <7 day post-fertilization (dpf) or three eyes of adult fish were collected and sonicated in RIPA Lysis Solution (Beyotime, Shanghai, China). The loading buffer was added and the protein samples were boiled for 10 min and then cooled on ice for 5 min. Protein concentration was measured with the BCA Protein Assay Kit (Beyotime, Shanghai, China). Western blot was performed as described previously [52]. The membranes were rinsed with SuperSignal® ELISA FemtoMaximum Sensitivity Substrate (Thermo Scientific, USA) and pictures were captured with ChemiDoc XRS + imaging system (Bio-Rad Laboratories). The list of antibodies used in the western blot is provided in Table S1.
Real-time fluorescence quantitative PCR (qRT-PCR)
Heads of the embryos were used for total RNA extraction. For each sample, 35 embryos were used. Total RNA was extracted with the RNA isolater Total RNA Extraction Reagent (Vazyme Biotech, Nanjing, China). The cDNA was synthesized using the HiScript II Q RT SuperMix for qPCR (+gDNA wiper) kit (Vazyme Biotech, Nanjing, China) according to the manufacturer’s manual. qRT-PCR was performed using the AceQ™ qPCR SYBR Green Master Mix (Vazyme Biotech, Nanjing, China) on the StepOnePlus™ real-time PCR system (Life Technologies). Data were analyzed with the GraphPad 5.1 software. Primers used for qRT-PCR are listed in Table S2.
Transmission electron microscopy (TEM)
For ultrastructural analysis, zebrafish embryos at 5 dpf were collected and processed as previously described [53]. The embryos were fixed in 2.5% glutaraldehyde (Servicebio, Wuhan, China) overnight at 4 °C. After three washes with PBS buffer, the embryos were further fixed in 1% osmium tetroxide for 2 h at RT. Next, the embryos were gradient dehydrated with ethanol and incubated in acetone for 20 min at RT. After treatment with propylene oxide, the embryos were embedded in epoxy resin. Then Reichert-Jung ultramicrotome was then used to produce ultrathin slices. Images were taken using the transmission electron microscope (HT7700, Hitachi, Japan).
Whole-mount in situ hybridization
Whole-mount in situ hybridization (WISH) was performed as previously described [54]. In brief, all DNA templates for the RNA probes were amplified from the cDNA library of whole embryos at 4 dpf, and cloned into the pGEM®-T Easy plasmid (Promega, A1360). The sequences of the in vitro transcription templates were validated by Sanger sequencing. The RNA probes were transcribed using the MAXIscript™ SP6/T7 Transcription Kit (Invitrogen, USA). The primer sequences used to amplify the DNA templates for synthesizing RNA probes were as follows: tulp1a: forward, AAAGAAGAAAGGCAAAGG; reverse, GTATCGGCACTCGCTCA. tulp1b: forward, AAAGAAAGCGGCCAAATCCGA; reverse, CGGGTCACTTTGCACTTCAC. tekt2: forward, GCCGGGCAAGGGAAGAGATG; reverse, AGCGCTGCCGGGTCTGGT.
Perls/DAB staining
The retinal frozen sections were treated with 30% hydrogen peroxide solution to remove pigment and soaked in PBS 3 times for 2 min each. The sections were then incubated with Perls reagent (5% potassium ferrocyanide (Sigma, P3289) and 5% HCl in ddH2O) for 1 h at 25 °C. After removing the Perls reagent, the sections were washed with ddH2O 3 times for 2 min each. Sections were treated with DAB solution (Servicebio, China) for 30 min to amplify the signals, and then washed with ddH2O 3 times for 5 min each. Finally, sections were mounted with 90% glycerol in PBS (PH 7.6) and imaged with optical microscope BX53.
Cell culture, plasmid construction, and Luciferase Reporter Assay
ZF4 cells were raised using DMEM-12 (Dulbecco’s Modified Eagle Medium/Nutrient Mixture F-12, Gibco, Grand Island, New York, USA) containing 10% fetal bovine serum (FBS, Biological Industries) at 28.5 °C in the presence of 5% CO2. The full-length coding sequence of tulp1a and tulp1b were separately subcloned into the pEGFP-C1 vector (Miaolingbio, P0134). The promotor region (2.1 kb) of tekt2 was subcloned into the pGL3-basic luciferase reporter vector. Primers are listed in Table S3. Plasmids were transfected into ZF4 cells with X-tremeGENE HP DNA Transfection Reagent (Roche) in OMEM (Opti-MEMI Reduced Serum Medium, Gibco). The medium was changed to DMEM-12 containing 10% FBS after 8 h incubation. After 36 h, the cells were harvested and the activity of luciferase was examined using the Dual Luciferase® Reporter Assay System (Promega, Madison, Wisconsin, USA). The experiments of HEK293, HEK293T, and ARPE-19 cells were performed as described previously [53, 55]. All cells were routinely tested for mycoplasma at regular intervals throughout the whole course of the study.
RNA-seq and bioinformatics analysis
The heads of zebrafish larvae at 4 dpf were dissected for RNA extraction. Nanodrop2000 was used to measure the concentration and purity of the RNA samples. Agarose gel electrophoresis and Agilent were used to estimate the integrity of RNA. RNA-Seq was performed on Illumina Novaseq 6000 platform by Majorbio (Shanghai, China). Sequencing data were mapped to the reference genomes (the zebrafish GRCz11 genome) using HISAT2 software [56, 57]. RSEM was used to obtain the Read Counts of each gene/transcript by comparing the results to the genome and the genome annotation file, and then TPM conversion was performed to obtain the standardized gene/transcript expression level [58]. Differentially expressed (DE) genes were determined by the R package DESeq2 using the following cut-off values: FC ≥ 2 and adjusted P-value ≤ 0.05. KEGG (Kyoto Encyclopedia of Genes and Genomes) pathway enrichment analysis was performed on DE genes using scripts written in R language with default parameters. The KEGG pathway is considered as significantly enriched if P-value ≤ 0.05. Gene Set Enrichment Analysis (GSEA) and Volcano analysis were performed using the online Hiplot (https://hiplot.com.cn/) [59].
Statistical analysis
All data were analyzed with a two-tailed t-test by using the GraphPad Prism 6 software and represented as mean ± SD. All the experiments were independently repeated at least three times. P-value was used to indicate significance: *P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001. n.s (no significance) P > 0.05.
Data availability
The NGS data are available under the GEO accession numbers: GSE214642
References
Khan AO, Bergmann C, Eisenberger T, Bolz HJ. A TULP1 founder mutation, p.Gln301*, underlies a recognisable congenital rod-cone dystrophy phenotype on the Arabian Peninsula. Br J Ophthalmol. 2015;99:488–92.
Banerjee P, Kleyn PW, Knowles JA, Lewis CA, Ross BM, Parano E, et al. TULP1 mutation in two extended Dominican kindreds with autosomal recessive retinitis pigmentosa. Nat Genet. 1998;18:177–9.
Hagstrom SA, North MA, Nishina PL, Berson EL, Dryja TP. Recessive mutations in the gene encoding the tubby-like protein TULP1 in patients with retinitis pigmentosa. Nat Genet. 1998;18:174–6.
den Hollander AI, van Lith-Verhoeven JJ, Arends ML, Strom TM, Cremers FP, Hoyng CB. Novel compound heterozygous TULP1 mutations in a family with severe early-onset retinitis pigmentosa. Arch Ophthalmol. 2007;125:932–5.
Mataftsi A, Schorderet DF, Chachoua L, Boussalah M, Nouri MT, Barthelmes D, et al. Novel TULP1 mutation causing leber congenital amaurosis or early onset retinal degeneration. Invest Ophthalmol Vis Sci. 2007;48:5160–7.
Abbasi AH, Garzozi HJ, Ben-Yosef T. A novel splice-site mutation of TULP1 underlies severe early-onset retinitis pigmentosa in a consanguineous Israeli Muslim Arab family. Mol Vis. 2008;14:675–82.
Jacobson SG, Cideciyan AV, Huang WC, Sumaroka A, Roman AJ, Schwartz SB, et al. TULP1 mutations causing early-onset retinal degeneration: preserved but insensitive macular cones. Invest Ophthalmol Vis Sci. 2014;55:5354–64.
Verbakel SK, Fadaie Z, Klevering BJ, van Genderen MM, Feenstra I, Cremers FPM, et al. The identification of a RNA splice variant in TULP1 in two siblings with early-onset photoreceptor dystrophy. Mol Genet Genom Med. 2019;7:e660.
Paloma E, Hjelmqvist L, Bayes M, Garcia-Sandoval B, Ayuso C, Balcells S, et al. Novel mutations in the TULP1 gene causing autosomal recessive retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2000;41:656–9.
Ikeda A, Nishina PM, Naggert JK. The tubby-like proteins, a family with roles in neuronal development and function. J Cell Sci. 2002;115:9–14.
North MA, Naggert JK, Yan Y, Noben-Trauth K, Nishina PM. Molecular characterization of TUB, TULP1, and TULP2, members of the novel tubby gene family and their possible relation to ocular diseases. Proc Natl Acad Sci USA. 1997;94:3128–33.
Boggon TJ, Shan WS, Santagata S, Myers SC, Shapiro L. Implication of tubby proteins as transcription factors by structure-based functional analysis. Science. 1999;286:2119–25.
Xi Q, Pauer GJ, Marmorstein AD, Crabb JW, Hagstrom SA. Tubby-like protein 1 (TULP1) interacts with F-actin in photoreceptor cells. Invest Ophthalmol Vis Sci. 2005;46:4754–61.
Milam AH, Hendrickson AE, Xiao M, Smith JE, Possin DE, John SK, et al. Localization of tubby-like protein 1 in developing and adult human retinas. Invest Ophthalmol Vis Sci. 2000;41:2352–6.
Hagstrom SA, Duyao M, North MA, Li T. Retinal degeneration in tulp1-/- mice: vesicular accumulation in the interphotoreceptor matrix. Invest Ophthalmol Vis Sci. 1999;40:2795–802.
Ikeda S, He W, Ikeda A, Naggert JK, North MA, Nishina PM. Cell-specific expression of tubby gene family members (tub, Tulp1,2, and 3) in the retina. Invest Ophthalmol Vis Sci. 1999;40:2706–12.
Hagstrom SA, Watson RF, Pauer GJ, Grossman GH. Tulp1 is involved in specific photoreceptor protein transport pathways. Adv Exp Med Biol. 2012;723:783–9.
Ran J, Zhou J. Targeting the photoreceptor cilium for the treatment of retinal diseases. Acta Pharm Sin. 2020;41:1410–5.
Ramamurthy V, Cayouette M. Development and disease of the photoreceptor cilium. Clin Genet. 2009;76:137–45.
Bachmann-Gagescu R, Neuhauss SC. The photoreceptor cilium and its diseases. Curr Opin Genet Dev. 2019;56:22–33.
Badgandi HB, Hwang SH, Shimada IS, Loriot E, Mukhopadhyay S. Tubby family proteins are adapters for ciliary trafficking of integral membrane proteins. J Cell Biol. 2017;216:743–60.
Hong JJ, Kim KE, Park SY, Bok J, Seo JT, Moon SJ. Differential roles of tubby family proteins in ciliary formation and trafficking. Mol Cells. 2021;44:591–601.
Remez L, Cohen B, Nevet MJ, Rizel L, Ben-Yosef T. TULP1 and TUB are required for specific localization of PRCD to photoreceptor outer segments. Int J Mol Sci. 2020;21:8677.
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–72.
Cui J, Zhao S, Li Y, Zhang D, Wang B, Xie J, et al. Regulated cell death: discovery, features and implications for neurodegenerative diseases. Cell Commun Signal. 2021;19:120.
Song D, Song Y, Hadziahmetovic M, Zhong Y, Dunaief JL. Systemic administration of the iron chelator deferiprone protects against light-induced photoreceptor degeneration in the mouse retina. Free Radic Biol Med. 2012;53:64–71.
Wang K, Peng B, Xiao J, Weinreb O, Youdim MBH, Lin B. Iron-chelating drugs enhance cone photoreceptor survival in a mouse model of retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2017;58:5287–97.
Chhetri J, Jacobson G, Gueven N. Zebrafish–on the move towards ophthalmological research. Eye. 2014;28:367–80.
Liu F, Qin Y, Huang Y, Gao P, Li J, Yu S, et al. Rod genesis driven by mafba in an nrl knockout zebrafish model with altered photoreceptor composition and progressive retinal degeneration. PLoS Genet. 2022;18:e1009841.
Kumaran N, Moore AT, Weleber RG, Michaelides M. Leber congenital amaurosis/early-onset severe retinal dystrophy: clinical features, molecular genetics and therapeutic interventions. Br J Ophthalmol. 2017;101:1147–54.
Durcan TM, Halpin ES, Rao T, Collins NS, Tribble EK, Hornick JE, et al. Tektin 2 is required for central spindle microtubule organization and the completion of cytokinesis. J Cell Biol. 2008;181:595–603.
Li X, Wang TX, Huang X, Li Y, Sun T, Zang S, et al. Targeting ferroptosis alleviates methionine-choline deficient (MCD)-diet induced NASH by suppressing liver lipotoxicity. Liver Int. 2020;40:1378–94.
Chen X, Li J, Kang R, Klionsky DJ, Tang D. Ferroptosis: machinery and regulation. Autophagy. 2021;17:2054–81.
Kubota A, Kawai YK, Yamashita N, Lee JS, Kondoh D, Zhang S, et al. Transcriptional profiling of cytochrome P450 genes in the liver of adult zebrafish, Danio rerio. J Toxicol Sci. 2019;44:347–56.
He X, Hahn P, Iacovelli J, Wong R, King C, Bhisitkul R, et al. Iron homeostasis and toxicity in retinal degeneration. Prog Retin Eye Res. 2007;26:649–73.
Chen C, Chen J, Wang Y, Liu Z, Wu Y. Ferroptosis drives photoreceptor degeneration in mice with defects in all-trans-retinal clearance. J Biol Chem. 2020.296:100187.
Fuhrmann DC, Mondorf A, Beifuss J, Jung M, Brune B. Hypoxia inhibits ferritinophagy, increases mitochondrial ferritin, and protects from ferroptosis. Redox Biol. 2020;36:101670.
Lu S, Wang XZ, He C, Wang L, Liang SP, Wang CC, et al. ATF3 contributes to brucine-triggered glioma cell ferroptosis via promotion of hydrogen peroxide and iron. Acta Pharmacol Sin. 2021.42:1690–1702
Santagata S, Boggon TJ, Baird CL, Gomez CA, Zhao J, Shan WS, et al. G-protein signaling through tubby proteins. Science. 2001;292:2041–50.
Alshawa E, Laqqan M, Montenarh M, Hammadeh ME. Influence of cryopreservation on the CATSPER2 and TEKT2 expression levels and protein levels in human spermatozoa. Toxicol Rep. 2019;6:819–24.
Ryan R, Failler M, Reilly ML, Garfa-Traore M, Delous M, Filhol E, et al. Functional characterization of tektin-1 in motile cilia and evidence for TEKT1 as a new candidate gene for motile ciliopathies. Hum Mol Genet. 2018;27:266–82.
Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, Stockwell BR. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci USA. 2016;113:E4966–4975.
Wang Z, Ding Y, Wang X, Lu S, Wang C, He C, et al. Pseudolaric acid B triggers ferroptosis in glioma cells via activation of Nox4 and inhibition of xCT. Cancer Lett. 2018;428:21–33.
Picard E, Daruich A, Youale J, Courtois Y, Behar-Cohen F. From rust to quantum biology: the role of iron in retina physiopathology. Cells. 2020;9:705.
Yefimova MG, Jeanny JC, Keller N, Sergeant C, Guillonneau X, Beaumont C, et al. Impaired retinal iron homeostasis associated with defective phagocytosis in Royal College of Surgeons rats. Invest Ophthalmol Vis Sci. 2002;43:537–45.
Caberoy NB, Zhou Y, Li W. Tubby and tubby-like protein 1 are new MerTK ligands for phagocytosis. EMBO J. 2010;29:3898–910.
Masek M, Etard C, Hofmann C, Hulsmeier AJ, Zang J, Takamiya M, et al. Loss of the Bardet-Biedl protein Bbs1 alters photoreceptor outer segment protein and lipid composition. Nat Commun. 2022;13:1282.
Stockwell BR. Ferroptosis turns 10: emerging mechanisms, physiological functions, and therapeutic applications. Cell. 2022;185:2401–21.
Labun K, Montague TG, Krause M, Torres Cleuren YN, Tjeldnes H, Valen E. CHOPCHOP v3: expanding the CRISPR web toolbox beyond genome editing. Nucleic Acids Res. 2019;47:W171–W174.
Qu Z, Yimer TA, Xie S, Wong F, Yu S, Liu X, et al. Knocking out lca5 in zebrafish causes cone-rod dystrophy due to impaired outer segment protein trafficking. Biochim Biophys Acta Mol Basis Dis. 2019;1865:2694–705.
Yu S, Li C, Biswas L, Hu X, Liu F, Reilly J, et al. CERKL gene knockout disturbs photoreceptor outer segment phagocytosis and causes rod-cone dystrophy in zebrafish. Hum Mol Genet. 2017;26:2335–45.
Liu F, Chen J, Yu S, Raghupathy RK, Liu X, Qin Y, et al. Knockout of RP2 decreases GRK1 and rod transducin subunits and leads to photoreceptor degeneration in zebrafish. Hum Mol Genet. 2015;24:4648–59.
Lu Z, Hu X, Reilly J, Jia D, Liu F, Yu S, et al. Deletion of the transmembrane protein Prom1b in zebrafish disrupts outer-segment morphogenesis and causes photoreceptor degeneration. J Biol Chem. 2019;294:13953–63.
Yu S, Jiang T, Jia D, Han Y, Liu F, Huang Y, et al. BCAS2 is essential for hematopoietic stem and progenitor cell maintenance during zebrafish embryogenesis. Blood. 2019;133:805–15.
Li J, Liu F, Lv Y, Sun K, Zhao Y, Reilly J, et al. Prpf31 is essential for the survival and differentiation of retinal progenitor cells by modulating alternative splicing. Nucleic Acids Res. 2021;49:2027–43.
Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36.
Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nat Methods. 2015;12:357–60.
Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinforma. 2011;12:323.
Li J, Miao B, Wang S, Dong W, Xu H, Si C, et al. Hiplot: a comprehensive and easy-to-use web service boosting the publication-ready biomedical data visualization. biorxiv. 2022.23:bbac261.
Acknowledgements
The authors thank Shanshan Yu (Wuhan University of Science and Technology), Zhaojing Lu (Zhongshan Ophthalmic Center), and Xuebin Hu (Zhongshan Ophthalmic Center) for their valuable advices.
Funding
This work was supported by the Ministry of Science and Technology of China (No. 2018YFA0801000), and the National Nature Science Foundation of China (Nos. 82071010, 31871260, and 81870691).
Author information
Authors and Affiliations
Contributions
ML, XR, ZT, and FL performed study concept; XR and DJ designed the study; DJ, PG, and YL performed most of the experiments in zebrafish and cell lines. XR, FL, and PG performed development of methodology; DJ, YL, FL, XS, ML, XR, and JR performed writing, review and revision of the paper; Y Han, KS, and Y Huang contributed to vector construction and frozen section; XC, ZZ, PL, and JL helped with the WISH experiments; DJ, PG, YL, YH, HH, FL, and ML provided acquisition, analysis and interpretation of data, and statistical analysis; ZT provided technical and material support. All authors read and approved the final paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics statement
All the procedures involving zebrafish were approved by the Ethics Committee of Huazhong University of Science and Technology.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by Professor Nicolas Baza
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Jia, D., Gao, P., Lv, Y. et al. Tulp1 deficiency causes early-onset retinal degeneration through affecting ciliogenesis and activating ferroptosis in zebrafish. Cell Death Dis 13, 962 (2022). https://doi.org/10.1038/s41419-022-05372-w
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-022-05372-w
