
Abstract
Systemic juvenile idiopathic arthritis (s-JIA) is clinically distinct from other types of JIA. It is typified by extraarticular features such as quotidian fevers, rash, splenomegaly, lymphadenopathy, laboratory abnormalities (including leukocytosis, thrombocytosis, anemia, hyperferritinemia, and elevated inflammatory markers), and a close association with the macrophage activation syndrome. Recent investigations have highlighted dysregulation of the innate immune system as the critical pathogenic driver of s-JIA. Key innate immune mediators of s-JIA are the macrophage-derived cytokines interleukin-1 (IL-1) and IL-6. Increased understanding of the roles of IL-1 and IL-6 in the pathogenesis of s-JIA has led to major changes in therapeutic options. Until recently, the most commonly used medications included corticosteroids, methotrexate, and tumor necrosis factor (TNF) inhibitors, which are incompletely effective in most cases. Newer biologic agents targeting IL-1 and IL-6 have proven very effective in treating s-JIA and in minimizing corticosteroid exposure. Here we review recent advances in the understanding of the pathogenesis of s-JIA and the recent clinical trials that have revolutionized the care of children with s-JIA.
Similar content being viewed by others


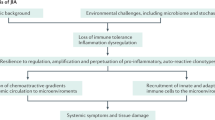
Clinical Characteristics of Systemic JIA
Systemic juvenile idiopathic arthritis (s-JIA) has unique features that differentiate it from other forms of JIA. Historically, s-JIA has been referred to as Still’s disease because it was first described in 1896 by George F. Still, who reported 22 children with chronic progressive enlargement of joints, general enlargement of lymph nodes, enlargement of the spleen, fever, and often anemia (1). The International League of Associations for Rheumatology (ILAR) has developed the following criteria to establish the diagnosis of s-JIA: arthritis in one or more joints with or preceded by fever of at least 2-wk duration that is documented to be daily (“quotidian”) for at least 3 d and is accompanied by one or more of the following: evanescent (nonfixed) erythematous rash, generalized lymph node enlargement, hepatomegaly and/or splenomegaly, and serositis. The ILAR criteria attempt to differentiate the various subtypes of JIA based on their primary clinical and laboratory characteristics to improve the homogeneity of research study subject enrollment and the resulting treatment strategies. Based on the ILAR criteria, exclusions to the diagnosis of s-JIA are features that are more typical of other subtypes of JIA, including psoriasis in the patient or a first-degree relative (suggesting psoriatic JIA); human leukocyte antigen B27 positivity in a male with arthritis starting after the age of 6 y (suggesting enthesitis-related JIA); ankylosing spondylitis, enthesitis-related arthritis, sacroiliitis with inflammatory bowel disease, Reiter’s syndrome, or acute anterior uveitis, or a history of one of these disorders in a first-degree relative (suggesting enthesitis-related JIA); or a positive immunoglobulin M (IgM) rheumatoid factor on two occasions at least 3 mo apart (suggesting rheumatoid factor–positive polyarticular JIA; see online Supplementary Appendix online) (2).
According to studies from European nations, s-JIA accounts for ~4–9% of all cases of JIA; however, the subjects in these studies were primarily Caucasian (3,4). In contrast, s-JIA comprises a much higher proportion of cases of JIA in Asian epidemiologic studies: e.g., a Japanese study conducted in 1994 revealed that s-JIA accounted for 54% of JIA cases (5). Additionally, early reports indicated that s-JIA comprised 24% of JIA cases in India (6), but more recent studies put this number at only 8%, similar to the proportion in most other countries (7). According to a population study conducted in Norway, the incidence of s-JIA is ~0.6 per 100,000/y (4). There is no sex bias, and the typical onset of symptoms is usually from 1 to 5 y of age, with a peak incidence at 2 y of age (8).
s-JIA is a clinical diagnosis, and several other mimicking conditions must be excluded. Malignancy, infection, and other rheumatic diseases can each cause fever, rash, and systemic inflammation. The most common clinical features of s-JIA include fever, arthritis, and rash, with at least 80% of patients presenting with these three features. Lymphadenopathy is a presenting sign in 30% of patients, and organomegaly and serositis are even less common. The arthritis can be polyarticular (>4 joints) or oligoarticular (≤4 joints) at presentation but is rarely monoarticular. The most commonly affected joints are the knees, wrists, and ankles, though any joint can be involved. Quotidian fevers are a classic feature of s-JIA, but only 64% of patients actually present with this fever pattern. Furthermore, according to one study, only 30% of patients with the clinical diagnosis of s-JIA actually fulfilled the ILAR criteria (8). Common laboratory abnormalities include evidence of systemic inflammation such as elevated erythrocyte sedimentation rate (mean 78 mm/h), C-reactive protein (CRP), white blood count (median 17.6 × 103 cells/μl), platelet count (median 539 × 103 cells/μl), and ferritin (usually >500 ng/ml). Mild anemia and elevated levels of transaminases, aldolase, and d-dimers are also commonly seen (8).
Historically, ~30% of patients with s-JIA eventually developed destructive polyarthritis (9) often requiring arthroplasty, and 40% of patients reported at least moderate functional disability (10).
Additionally, the pattern of the disease course is variable. Approximately 40% of children with s-JIA will follow a monophasic pattern and undergo remission after a single exacerbation, which may last up to 2 y. Approximately 7% will follow a polycyclic pattern in which patients experience episodes of disease exacerbation followed by periods of remission. Finally, up to 50% of patients historically experienced a persistent pattern, which is defined as persistent disease for at least 2 y. Predictors of nonmonophasic disease patterns include fevers and active arthritis at 3 mo and elevated erythrocyte sedimentation rate (>26 mm/h) and corticosteroid use at 6 mo (11). With the advent of newer biologic therapeutic agents discussed below, disease outcomes are expected to improve dramatically.
Innate Immune System
Recent evidence supports the idea that s-JIA arises due to dysregulation of the innate immune system. This is in contrast with other forms of JIA, which are driven primarily by the adaptive immune system. Dysregulation of the innate immune system in s-JIA results in increased production of inflammatory cytokines, leading to the distinctive clinical features of the disease. A recent study compared gene expression profiles among patients with different subtypes of JIA. Genes that were found to be uniquely upregulated in s-JIA compared with other subtypes included innate immune pathways (interleukin-6 (IL-6), toll-like receptor/IL-1 receptor, and peroxisome proliferator–activated receptor-γ (PPARγ) signaling). Genes related to natural killer cells and T-cells were downregulated (12). This recent understanding of the pathogenesis of s-JIA and the predominant role of the innate immune system has led to new therapeutic options that focus on inhibiting the effects of key proinflammatory cytokines.
Cytokines
The macrophage-derived cytokines IL-6, IL-1, and IL-18 predominate the inflammatory milieu in s-JIA (13). Gene expression profiles have shown increased expression of IL-6 in blood monocytes (and B-cells) in patients with active s-JIA compared with the expression in healthy controls (14). Additionally, serum IL-6 levels are significantly elevated in s-JIA patients compared with the same in healthy, age-matched controls. IL-6 levels correlate significantly with the extent and severity of joint symptoms and platelet count levels, and IL-6 levels decrease as patients enter remission (15). Studies in humans and animals have indicated that chronically elevated IL-6 levels can lead to both microcytic anemia and impaired growth, common findings among patients with s-JIA (16,17,18).
IL-1β is a key contributor to the pathogenesis of s-JIA. Activated peripheral blood mononuclear cells from patients with s-JIA produce large amounts of IL-1β. Furthermore, serums from patients with s-JIA can induce transcription of innate immune genes, including the IL-1 locus, in peripheral blood mononuclear cells of healthy controls. In this early study of nine patients with active s-JIA treated with recombinant IL-1 receptor antagonist, all of the subjects experienced a partial or complete response to treatment, further supporting the idea that IL-1 plays a key pathogenic role (19). This study contrasts slightly with others that did not find increased gene expression of IL-1β in the peripheral blood mononuclear cells of patients with s-JIA but did find upregulation of IL-1-initiated signaling pathways (14,20). Although the exact cellular source of IL-1 in s-JIA remains an open question, the clinical experience with IL-1-blocking agents, discussed below, confirms the important role of IL-1 in disease pathogenesis.
Cytokine profile studies have suggested that IL-18 levels also correspond to disease activity status of s-JIA (13). IL-18 levels in both serum and synovial fluid positively correlate with CRP level, the number of active joints, radiological score, and levels of IL-6, IL-1, and IL-1 receptor antagonist. Although IL-18 levels were elevated in children with active JIA of all subtypes, these levels were markedly higher in the synovial fluid and serums of those with s-JIA compared with those children with the other JIA subtypes (21). A recent study evaluated whether differences in clinical features among s-JIA patients could be correlated with a predominance of IL-6 vs. IL-18. This study found that patients with IL-6-predominant disease were more likely to have a greater number of active joints, whereas those with IL-18-predominant disease were more likely to develop macrophage activation syndrome (MAS) (22).
Thus, macrophage-derived cytokines are more than simply markers of disease activity and appear to be key players in the pathogenesis of s-JIA. This recognition has increased our understanding of the complications that can arise in s-JIA and has led to exciting new therapeutic approaches.
Macrophage Activation Syndrome
MAS is a form of hemophagocytic lymphohistiocytosis that occurs in children with rheumatic diseases. MAS is associated with s-JIA and less commonly with other rheumatic diseases in children (23). The clinical features of MAS include fever, hepatosplenomegaly, lymphadenopathy, cytopenias, coagulopathy, and central nervous system inflammation. It can also be associated with rash, serositis, myositis, renal failure, and respiratory failure. Laboratory findings include hyperferritinemia (often marked), elevated d-dimer level and prothrombin time, elevated liver enzymes, and decreased fibrinogen and cell counts (23). The most commonly reported triggers of MAS are infections and changes in medications, with Epstein–Barr virus being the most common etiology (24). Morbidity and mortality among patients with MAS is high, with one study reporting a mortality rate of 22% (25). MAS is thought to be caused by uncontrolled and persistent expansion of cytotoxic lymphocytes that produce inflammatory cytokines, which then induce activation of hemophagocytic (CD163+) macrophages (26). Early reports suggested that MAS occurs in ~7% of patients with s-JIA (25);however, more recent studies have suggested that occult MAS is much more common in children with s-JIA than previously recognized. One such study analyzed the bone marrow of 15 patients with a known diagnosis of s-JIA and found that 8 of them (53%) had histopathologic findings consistent with MAS, but only 2 of them had clinically diagnosed MAS. There were no significant differences in the laboratory values of those patients with s-JIA who did and did not have evidence of MAS in the bone marrow (27).
Soluble IL-2 receptor α (sIL-2Rα, sCD25) has been found to correlate with the degree of T-cell activation, and soluble CD163 (sCD163) correlates with the degree of macrophage activation. The levels of soluble IL-2Rα and sCD163 were determined in 7 patients with new-onset s-JIA and 16 patients with MAS. Median levels of sIL-2Rα and sCD163 were significantly higher in subjects with MAS compared with those with new-onset s-JIA; however, in 5 of the 16 patients with s-JIA, the sIL-2Rα and sCD163 levels were similar to those with MAS. These five patients were also noted to have higher disease activity, with higher levels of hemoglobin and serum ferritin and lower platelet counts than the other patients with s-JIA; two of these five patients developed MAS within 1 mo (26). These studies support the concept that s-JIA and MAS represent a spectrum of disease with common pathogenic mechanisms. Further support of this notion comes from genetic studies in patients with s-JIA and MAS. One form of familial hemophagocytic lymphohistiocytosis (FHL3) arises due to mutations in the gene encoding Munc13-4 (UNC13D), a protein involved in cytotoxic lymphocyte granule exocytosis. An initial case report highlighted a girl with s-JIA without MAS who was found to have compound heterozygous mutations of UNC13D (28). A larger study assessed UNC13D sequence alterations among patients with s-JIA and MAS. This study determined that 2 of 18 patients with s-JIA and MAS had a mutation in the UNC13D gene. Furthermore, 9 (56%) of the remaining 16 patients with s-JIA and MAS had an identical combination of 12 single-nucleotide polymorphisms in the UNC13D gene compared with 12% of healthy controls and 8.2% of patients with s-JIA and without MAS (29). These studies, in concert with other studies demonstrating decreased natural killer cell function and decreased expression of perforin protein (encoded by the gene responsible for FHL2) among patients with s-JIA, suggest that s-JIA/MAS and hemophagocytic lymphohistiocytosis are pathogenically related (30).
High-dose i.v. methylprednisolone is the usual initial treatment for MAS (31). Cyclosporine A is another important therapeutic agent, particularly for individuals unresponsive to corticosteroids (32). Etoposide may also be used to treat MAS but is generally reserved for those who fail corticosteroid and cyclosporine A therapy as this treatment is associated with serious toxicity, including bone marrow suppression and serious infections (33). An alternative therapy to etoposide is antithymocyte globulin; however, this treatment is also less commonly used in those with MAS (34). More recently, the IL-1 blocking agent, anakinra, has been successfully used to treat MAS (35); however, it should be noted that some patients with s-JIA have developed MAS while being managed with anakinra (36). Immunoglobulin (i.v.) and rituximab have also been used successfully, particularly in the setting of MAS triggered by Epstein–Barr virus (37,38). Greater detail regarding treatment strategies for MAS can be found in the HLH-2004 therapeutic guidelines (39).
Therapy
The new advances in understanding the pathogenesis of s-JIA have revolutionized therapy for affected children. Until recently, the mainstay of therapy consisted of corticosteroids and disease-modifying antirheumatic drugs with the goal of sparing corticosteroid use. One of the most commonly used disease-modifying antirheumatic drugs is methotrexate. Although methotrexate monotherapy has been shown to be beneficial for other types of JIA, no significant overall improvement was noted among s-JIA patients treated with methotrexate in a randomized, placebo-controlled crossover study (40). Other therapies that have been tried with variable success and with limited available data include cyclosporine A (41,42), i.v. immunoglobulin (43,44), thalidomide (45), and intraarticular triamcinolone hexacetonide (46).
Anti-tumor necrosis factor (TNF) agents also have poor response rates among patients with s-JIA, especially compared with their effectiveness in other JIA subtypes. A prospective cohort study conducted in 2009 evaluated the effectiveness of anti-TNF agents among 45 patients with s-JIA. The study showed that 24% of patients entered remission, but remission was sustained in only 13%. The TNF agents evaluated included etanercept (Amgen, Thousand Oaks, CA), infliximab (Janssen Biotech, Horsham, PA), and adalimumab (AbbVie North Chicago, IL) (47). Thankfully, greater success has been achieved with newer biologic agents, particularly those targeting IL-1 and IL-6 ( Table 1 ). Current therapeutic options for other JIA subtypes have recently been reviewed elsewhere (48).
IL-1 blockade
The IL-1 inhibitor, anakinra (Amgen), was the first biologic medication to revolutionize treatment of s-JIA. Anakinra is a recombinant IL-1 receptor antagonist approved by the US Food and Drug Administration for the treatment of active rheumatoid arthritis in adults and autoinflammatory cryopyrin-associated periodic syndromes, including Muckle–Wells syndrome, familial cold autoinflammatory syndrome, and neonatal-onset multisystem inflammatory disease (49,50,51). The effectiveness of anakinra in s-JIA was first reported in 2004 in two patients who had refractory disease despite treatment with corticosteroids, methotrexate, and various TNF-inhibitors. Both patients responded quickly and fully to anakinra (52).
In 2011, two larger and more comprehensive studies reported the efficacy of anakinra in s-JIA. The first was a multicenter, double-blind, placebo-controlled trial including 24 patients. The primary objective of this study was to compare the efficacy of 1 mo of treatment with anakinra vs. placebo. Each group included 12 patients, and efficacy was defined as a 30% improvement in criteria of American College of Rheumatology for pediatric response (ACR Pedi 30) (53), resolution of systemic symptoms, and at least a 50% decrease in CRP and erythrocyte sedimentation rate. After 1 mo, there was a significant difference in response rates, with 67% (8/12) of anakinra-treated subjects achieving the primary end point compared with 8% (1/12) in the placebo group. Gene expression profiling demonstrated normalization of previously dysregulated genes and upregulation of interferon (IFN)-inducible genes among the anakinra-treated subjects (54). The second study was a multicenter international observational trial analyzing 46 s-JIA patients treated with anakinra. The primary goal of this study was to analyze the safety and efficacy of anakinra as first-line therapy in the early disease phase (in contrast with other studies in which anakinra was introduced after multiple other therapies had been incompletely effective). This study showed resolution of systemic symptoms and prevention of refractory arthritis in 90% of the study participants. In addition, at 2 mo, the majority of subjects no longer required corticosteroids (36). These studies highlight the effectiveness of IL-1 antagonism with anakinra in treating s-JIA. A practical drawback of anakinra is the need for daily s.c. injections, prompting the investigation of other IL-1-blocking agents in s-JIA.
A newer IL-1 inhibitor used to treat s-JIA is canakinumab (Novartis International AG, Basel, Switzerland), a fully humanized monoclonal antibody that binds selectively to IL-1β, inactivating its signaling. Similar to anakinra, canakinumab is administered by s.c. injection, but it has a longer half-life and thus needs to be given only once monthly (55). A recent report described two phase III trials that demonstrated the effectiveness of canakinumab in treating s-JIA. The first trial was a double-blind, placebo-controlled study with a primary outcome measure of adapted JIA ACR 30 response with resolution of fever within 15 d of receiving the medication; in this trial, 33% of participants achieved inactive disease within 15 d of the first dose of canakinumab. Furthermore, 84% of the treatment group achieved an ACR 30 response compared with 10% in the placebo group. These statistically significant results support the notion that canakinumab is effective and produces a rapid clinical response. The second trial was a two-phase trial. The first phase was an open-label one, in which all subjects received canakinumab with the goal of tapering corticosteroids. Forty-five percent of patients on canakinumab were able to taper their corticosteroid doses, and 33% of the patients were able to discontinue corticosteroids altogether. The second phase was a double-blind, placebo-controlled withdrawal study with the primary outcome of time to flare. Seventy-five percent of the subjects in the canakinumab group did not have a flare, whereas only 25% of the subjects in the placebo group did not have a flare, a statistically significant difference. Infections were more common among subjects receiving canakinumab. Additionally, neutropenia and thrombocytopenia were noted; however, in most cases, this occurrence was transient and resolved without intervention (56).
Rilonacept (“IL-1 trap”; Regeneron Pharmaceuticals, Tarrytown, NY) is another IL-1 antagonist that might prove useful in s-JIA. It is a fusion molecule composed of the extracellular component of the IL-1 receptor and the Fc portion of the IgG1, which binds circulating IL-1β and IL-1α with high affinity. Rilonacept was first shown to be safe and effective in the treatment of cryopyrin-associated periodic syndromes in 2008. The first study was a small pilot study including five patients with familial cold autoinflammatory syndrome and who had significant reduction in erythrocyte sedimentation rate, CRP, and serum amyloid A levels while on rilonacept (57). The second study was a larger two-step placebo-controlled trial evaluating the response of 47 patients with cryopyrin-associated periodic syndromes (including familial cold autoinflammatory syndrome and Muckle–Wells syndrome). In this study, rilonacept significantly reduced symptom scores, CRP and serum amyloid A levels, and the physician and patient global assessment of disease compared with placebo. Rilonacept was well tolerated, with the most common adverse effect being injection site reactions (58). A more recent, small, placebo-controlled study also found rilonacept promising in the treatment of Familial Mediterranean Fever, another genetic autoinflammatory syndrome in which IL-1 is an important mediator of disease (59). Although to date there have not yet been any published results from randomized controlled trials of rilonacept in s-JIA, a small case series showed promising results in the treatment of three patients with refractory adult-onset Still’s disease. All three patients in this series had inadequate control of disease on anakinra treatment but responded favorably to rilonacept (60). Additionally, preliminary data reported from a double-blind placebo-controlled trial of rilonacept in children with s-JIA suggest promising results; however, the study is not yet complete (61).
IL-6 Blockade
Tocilizumab (Genentech, South San Francisco, CA) is a humanized anti-human IL-6 receptor antibody that blocks both membrane-bound and soluble receptors. The first studies showing efficacy of tocilizumab in s-JIA were published in 2005. The first study was a small study that showed rapidly reduced disease activity in 10 of 11 Japanese children with active s-JIA (62). The second study was a small open-label single-dose phase II trial of 18 Caucasian children with s-JIA. This study found that 11 of the 18 subjects had at least a 30% improvement in symptoms within 48 h of a single dose, and this improvement was sustained for up to 8 wk (63). In 2008, a larger, double-blind, placebo-controlled phase III trial including 56 children with s-JIA showed the efficacy and safety of tocilizumab. The study began with a 6-wk open-label lead-in phase. Fifty-one (91%) of the 56 children in the open-label lead-in phase achieved at least an ACR Pedi 30 response, and 38 patients (68%) achieved ACR Pedi 70 responses. Subjects who achieved at least an ACR Pedi 30 response and a CRP level less than 5 mg/l were able to enroll in a 12-wk double-blind, placebo-controlled, randomized withdrawal phase. Forty-three subjects entered the double-blind phase of the trial in which the primary end point was ACR Pedi 30 response and CRP less than 15 mg/l. Eighty percent (18/20) of those in the tocilizumab group compared with 17% (4/23) of those in the placebo group maintained their ACR Pedi 30 response and CRP level less than 15 mg/l. The most common adverse events were mild gastroenteritis and nasopharyngeal and upper respiratory infections. Elevated transaminases were also noted at the initiation of tocilizumab but gradually decreased as treatment continued (64). A more recent randomized, placebo-controlled study evaluated the safety and efficacy of tocilizumab among 112 children who had had s-JIA for at least 6 mo and whose disease was refractory to nonsteroidal anti-inflammatory drugs and corticosteroids. The primary end point was absence of fever and ACR Pedi 30 response at the end of 12 wk of treatment. Eighty-five percent (64/75) of those in the tocilizumab group compared with 24% (9/37) of those in the placebo group achieved the primary end point. The patients then entered an open-label phase, and at 1 y, 80% of the participants had achieved at least an ACR Pedi 70 response without fever, 59% had at least an ACR Pedi 90 response, 48% had no joints with active arthritis, and 52% were able to discontinue corticosteroids. Similar to the previous study, the most common adverse events related to tocilizumab were infections, neutropenia, and elevated transaminases (65).
Consensus Treatment Plans
With the advent of these new biologic agents, many therapeutic options exist for patients with s-JIA. In 2012, the Childhood Arthritis and Rheumatology Research Alliance published consensus treatment plans and standardized assessment schedules to begin to understand the comparative effectiveness of the available treatment options in s-JIA. The four standardized treatment plans developed for the treatment of new-onset s-JIA included glucocorticoids only or treatment with methotrexate, anakinra, or tocilizumab, each with optional glucocorticoid use. Implementing these treatment plans and standardized assessment schedules is designed to allow for the evaluation of each treatment regimen’s comparative effectiveness (66). Data from the utilization of the consensus treatment plans continue to be collected at this time. It should be noted that these consensus treatment plans were developed before the publication of the results of the recent phase III trials of IL-1- and IL-6-blocking agents mentioned above (56,64), and that the consensus treatment plans do not specifically include canakinumab. In view of the successful results of those trials, it is likely that for patients with active systemic disease, most pediatric rheumatologists will recommend treatment regimens that include IL-1- or IL-6-blocking agents relative to those that include only corticosteroids and/or methotrexate. It should also be highlighted that despite the great improvement in s-JIA management with these new agents, they do not offer full protection against MAS. According to one study, up to 20% of patients receiving anakinra developed MAS (36), whereas three patients were reported to develop MAS while receiving tocilizumab therapy (65).
Other Therapeutic Strategies
Abatacept (Bristol-Myers Squibb, Princeton, NJ) is a soluble fusion protein consisting of the extracellular domain of human cytotoxic T lymphocyte–associated antigen 4 (CTLA-4) linked to a modified Fc portion of human IgG1. It is the first in a class of agents that selectively modulate the CD80/CD86:CD28 costimulatory signals needed for full T-cell activation (67). Abatacept has been shown to be beneficial in some cases of s-JIA; however, the majority of the subjects in this retrospective study had arthritis but not active systemic features (e.g., fever, rash). Because the mechanism of action (costimulatory molecule blockade) is different from that of the other biologic agents used to treat s-JIA (cytokine blockade), some investigators feel that abatacept may be safely used in combination with other biologics in patients with severe s-JIA (68). Further studies evaluating the efficacy of abatacept in certain clinical subgroups of s-JIA patients would be helpful.
Lastly, autologous T-cell depleted hematopoietic stem cell transplant has been used in the treatment of s-JIA since 1997. This approach is typically reserved for those patients who have an inadequate response to aggressive treatment with biologic and other therapies. Although patients may experience marked improvement or complete remission after autologous stem cell transplantation, there are real risks of severe morbidity and mortality (69). In a study evaluating the safety and efficacy of autologous stem cell transplantation in 22 children with severe and progressive systemic or polyarticular JIA, 15 of the 22 patients had complete or significant response to therapy. However, generally, the recovery of CD4+/CD45RA+-naïve T-cells was prolonged, and patients experienced many viral infections. These viral infections may have contributed to the development of MAS, resulting in the death of two patients. In response to these concerns, the protocol was changed to improve T-cell recovery; no additional deaths occurred after this change. Two additional patients died later due to infections attributed to their ongoing immunosuppressive therapy, and five patients experienced relapse of s-JIA disease (70). A second study published in 2009 from the United Kingdom evaluated seven children with s-JIA who also received T-cell depleted autologous stem cell transplant. Four of the seven children had a marked and sustained response (followed for up to 8 y) and were able to discontinue anti-inflammatory and immunosuppressive medications; however, one patient died within months after transplantation and two patients relapsed (71). With the advent of newer, more effective biologic therapies, today only rare patients with s-JIA are considered to be candidates for autologous stem cell transplantation.
Conclusion
In conclusion, recent studies have demonstrated that s-JIA is a disease stemming from dysregulation of the innate immune system characterized by production of macrophage-derived proinflammatory cytokines, namely IL-1, IL-6, and IL-18. Additional investigations have highlighted the potential links between s-JIA and hemophagocytic lymphohistiocytosis. Understanding the pathogenic mechanisms underlying s-JIA has generated great interest in newer biologic agents designed to block IL-1 and IL-6, which have revolutionized the treatment of s-JIA. As additional mechanistic insight is generated regarding the pathogenesis of s-JIA, we anticipate that additional therapeutic targets will emerge, further increasing the likelihood of inducing disease remission in children who suffer from this disease.
Statement of Financial Support
No extramural funds supported this work.
Disclosures:
The authors declare no conflict of interest.
References
Still GF . On a Form of Chronic Joint Disease in Children. Med Chir Trans 1897;80:47–60.9.
Petty RE, Southwood TR, Manners P, et al.; International League of Associations for Rheumatology. International League of Associations for Rheumatology classification of juvenile idiopathic arthritis: second revision, Edmonton, 2001. J Rheumatol 2004;31:390–2.
Danner S, Sordet C, Terzic J, et al. Epidemiology of juvenile idiopathic arthritis in Alsace, France. J Rheumatol 2006;33:1377–81.
Berntson L, Andersson Gäre B, Fasth A, et al.; Nordic Study Group. Incidence of juvenile idiopathic arthritis in the Nordic countries. A population based study with special reference to the validity of the ILAR and EULAR criteria. J Rheumatol 2003;30:2275–82.
Fujikawa S, Okuni M . Clinical analysis of 570 cases with juvenile rheumatoid arthritis: results of a nationwide retrospective survey in Japan. Acta Paediatr Jpn 1997;39:245–9.
Seth V, Kabra SK, Semwal OP, Jain Y . Clinico-immunological profile in juvenile rheumatoid arthritis–an Indian experience. Indian J Pediatr 1996;63:293–300.
Kunjir V, Venugopalan A, Chopra A . Profile of Indian patients with juvenile onset chronic inflammatory joint disease using the ILAR classification criteria for JIA: a community-based cohort study. J Rheumatol 2010;37:1756–62.
Behrens EM, Beukelman T, Gallo L, et al. Evaluation of the presentation of systemic onset juvenile rheumatoid arthritis: data from the Pennsylvania Systemic Onset Juvenile Arthritis Registry (PASOJAR). J Rheumatol 2008;35:343–8.
Schneider R, Lang BA, Reilly BJ, et al. Prognostic indicators of joint destruction in systemic-onset juvenile rheumatoid arthritis. J Pediatr 1992;120(2 Pt 1):200–5.
Oen K, Malleson PN, Cabral DA, Rosenberg AM, Petty RE, Cheang M . Disease course and outcome of juvenile rheumatoid arthritis in a multicenter cohort. J Rheumatol 2002;29:1989–99.
Singh-Grewal D, Schneider R, Bayer N, Feldman BM . Predictors of disease course and remission in systemic juvenile idiopathic arthritis: significance of early clinical and laboratory features. Arthritis Rheum 2006;54:1595–601.
Barnes MG, Grom AA, Thompson SD, et al. Subtype-specific peripheral blood gene expression profiles in recent-onset juvenile idiopathic arthritis. Arthritis Rheum 2009;60:2102–12.
de Jager W, Hoppenreijs EP, Wulffraat NM, Wedderburn LR, Kuis W, Prakken BJ . Blood and synovial fluid cytokine signatures in patients with juvenile idiopathic arthritis: a cross-sectional study. Ann Rheum Dis 2007;66:589–98.
Ogilvie EM, Khan A, Hubank M, Kellam P, Woo P . Specific gene expression profiles in systemic juvenile idiopathic arthritis. Arthritis Rheum 2007;56:1954–65.
de Benedetti F, Massa M, Robbioni P, Ravelli A, Burgio GR, Martini A . Correlation of serum interleukin-6 levels with joint involvement and thrombocytosis in systemic juvenile rheumatoid arthritis. Arthritis Rheum 1991;34:1158–63.
Cazzola M, Ponchio L, de Benedetti F, et al. Defective iron supply for erythropoiesis and adequate endogenous erythropoietin production in the anemia associated with systemic-onset juvenile chronic arthritis. Blood 1996;87:4824–30.
De Benedetti F, Rucci N, Del Fattore A, et al. Impaired skeletal development in interleukin-6-transgenic mice: a model for the impact of chronic inflammation on the growing skeletal system. Arthritis Rheum 2006;54:3551–63.
De Benedetti F, Meazza C, Oliveri M, et al. Effect of IL-6 on IGF binding protein-3: a study in IL-6 transgenic mice and in patients with systemic juvenile idiopathic arthritis. Endocrinology 2001;142:4818–26.
Pascual V, Allantaz F, Arce E, Punaro M, Banchereau J . Role of interleukin-1 (IL-1) in the pathogenesis of systemic onset juvenile idiopathic arthritis and clinical response to IL-1 blockade. J Exp Med 2005;201:1479–86.
Fall N, Barnes M, Thornton S, et al. Gene expression profiling of peripheral blood from patients with untreated new-onset systemic juvenile idiopathic arthritis reveals molecular heterogeneity that may predict macrophage activation syndrome. Arthritis Rheum 2007;56:3793–804.
Lotito AP, Campa A, Silva CA, Kiss MH, Mello SB . Interleukin 18 as a marker of disease activity and severity in patients with juvenile idiopathic arthritis. J Rheumatol 2007;34:823–30.
Shimizu M, Nakagishi Y, Yachie A . Distinct subsets of patients with systemic juvenile idiopathic arthritis based on their cytokine profiles. Cytokine 2013;61:345–8.
Hadchouel M, Prieur AM, Griscelli C . Acute hemorrhagic, hepatic, and neurologic manifestations in juvenile rheumatoid arthritis: possible relationship to drugs or infection. J Pediatr 1985;106:561–6.
Stéphan JL, Koné-Paut I, Galambrun C, Mouy R, Bader-Meunier B, Prieur AM . Reactive haemophagocytic syndrome in children with inflammatory disorders. A retrospective study of 24 patients. Rheumatology (Oxford) 2001;40:1285–92.
Sawhney S, Woo P, Murray KJ . Macrophage activation syndrome: a potentially fatal complication of rheumatic disorders. Arch Dis Child 2001;85:421–6.
Bleesing J, Prada A, Siegel DM, et al. The diagnostic significance of soluble CD163 and soluble interleukin-2 receptor alpha-chain in macrophage activation syndrome and untreated new-onset systemic juvenile idiopathic arthritis. Arthritis Rheum 2007;56:965–71.
Behrens EM, Beukelman T, Paessler M, Cron RQ . Occult macrophage activation syndrome in patients with systemic juvenile idiopathic arthritis. J Rheumatol 2007;34:1133–8.
Hazen MM, Woodward AL, Hofmann I, et al. Mutations of the hemophagocytic lymphohistiocytosis-associated gene UNC13D in a patient with systemic juvenile idiopathic arthritis. Arthritis Rheum 2008;58:567–70.
Zhang K, Biroschak J, Glass DN, et al. Macrophage activation syndrome in patients with systemic juvenile idiopathic arthritis is associated with MUNC13-4 polymorphisms. Arthritis Rheum 2008;58:2892–6.
Grom AA, Villanueva J, Lee S, Goldmuntz EA, Passo MH, Filipovich A . Natural killer cell dysfunction in patients with systemic-onset juvenile rheumatoid arthritis and macrophage activation syndrome. J Pediatr 2003;142:292–6.
Stéphan JL, Zeller J, Hubert P, Herbelin C, Dayer JM, Prieur AM . Macrophage activation syndrome and rheumatic disease in childhood: a report of four new cases. Clin Exp Rheumatol 1993;11:451–6.
Mouy R, Stephan JL, Pillet P, Haddad E, Hubert P, Prieur AM . Efficacy of cyclosporine A in the treatment of macrophage activation syndrome in juvenile arthritis: report of five cases. J Pediatr 1996;129:750–4.
Gupta AA, Tyrrell P, Valani R, Benseler S, Abdelhaleem M, Weitzman S . Experience with hemophagocytic lymphohistiocytosis/macrophage activation syndrome at a single institution. J Pediatr Hematol Oncol 2009;31:81–4.
Coca A, Bundy KW, Marston B, Huggins J, Looney RJ . Macrophage activation syndrome: serological markers and treatment with anti-thymocyte globulin. Clin Immunol 2009;132:10–8.
Bruck N, Suttorp M, Kabus M, Heubner G, Gahr M, Pessler F . Rapid and sustained remission of systemic juvenile idiopathic arthritis-associated macrophage activation syndrome through treatment with anakinra and corticosteroids. J Clin Rheumatol 2011;17:23–7.
Nigrovic PA, Mannion M, Prince FH, et al. Anakinra as first-line disease-modifying therapy in systemic juvenile idiopathic arthritis: report of forty-six patients from an international multicenter series. Arthritis Rheum 2011;63:545–55.
Larroche C, Bruneel F, André MH, et al.; Comité d’Evaluation et de Diffusion des Innovation Technologiques (CEDIT). [Intravenously administered gamma-globulins in reactive hemaphagocytic syndrome. Multicenter study to assess their importance, by the immunoglobulins group of experts of CEDIT of the AP-HP]. Ann Med Interne (Paris) 2000;151:533–9.
Balamuth NJ, Nichols KE, Paessler M, Teachey DT . Use of rituximab in conjunction with immunosuppressive chemotherapy as a novel therapy for Epstein Barr virus-associated hemophagocytic lymphohistiocytosis. J Pediatr Hematol Oncol 2007;29:569–73.
Henter JI, Horne A, Aricó M, et al. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer 2007;48:124–31.
Woo P, Southwood TR, Prieur AM, et al. Randomized, placebo-controlled, crossover trial of low-dose oral methotrexate in children with extended oligoarticular or systemic arthritis. Arthritis Rheum 2000;43:1849–57.
Gerloni V, Cimaz R, Gattinara M, Arnoldi C, Pontikaki I, Fantini F . Efficacy and safety profile of cyclosporin A in the treatment of juvenile chronic (idiopathic) arthritis. Results of a 10-year prospective study. Rheumatology (Oxford) 2001;40:907–13.
Ruperto N, Ravelli A, Castell E, et al.; Pediatric Rheumatology Collaborative Study Group (PRCSG); Paediatric Rheumatology International Trials Organisation (PRINTO). Cyclosporine A in juvenile idiopathic arthritis. Results of the PRCSG/PRINTO phase IV post marketing surveillance study. Clin Exp Rheumatol 2006;24:599–605.
Silverman ED, Cawkwell GD, Lovell DJ, et al. Intravenous immunoglobulin in the treatment of systemic juvenile rheumatoid arthritis: a randomized placebo controlled trial. Pediatric Rheumatology Collaborative Study Group. J Rheumatol 1994;21:2353–8.
Uziel Y, Laxer RM, Schneider R, Silverman ED . Intravenous immunoglobulin therapy in systemic onset juvenile rheumatoid arthritis: a followup study. J Rheumatol 1996;23:910–8.
Lehman TJ, Schechter SJ, Sundel RP, Oliveira SK, Huttenlocher A, Onel KB . Thalidomide for severe systemic onset juvenile rheumatoid arthritis: A multicenter study. J Pediatr 2004;145:856–7.
Breit W, Frosch M, Meyer U, Heinecke A, Ganser G . A subgroup-specific evaluation of the efficacy of intraarticular triamcinolone hexacetonide in juvenile chronic arthritis. J Rheumatol 2000;27:2696–702.
Russo RA, Katsicas MM . Clinical remission in patients with systemic juvenile idiopathic arthritis treated with anti-tumor necrosis factor agents. J Rheumatol 2009;36:1078–82.
Espinosa M, Gottlieb BS . Juvenile idiopathic arthritis. Pediatr Rev 2012;33:303–13.
Hawkins PN, Lachmann HJ, Aganna E, McDermott MF . Spectrum of clinical features in Muckle-Wells syndrome and response to anakinra. Arthritis Rheum 2004;50:607–12.
Goldbach-Mansky R, Dailey NJ, Canna SW, et al. Neonatal-onset multisystem inflammatory disease responsive to interleukin-1beta inhibition. N Engl J Med 2006;355:581–92.
Hoffman HM, Rosengren S, Boyle DL, et al. Prevention of cold-associated acute inflammation in familial cold autoinflammatory syndrome by interleukin-1 receptor antagonist. Lancet 2004;364:1779–85.
Verbsky JW, White AJ . Effective use of the recombinant interleukin 1 receptor antagonist anakinra in therapy resistant systemic onset juvenile rheumatoid arthritis. J Rheumatol 2004;31:2071–5.
Giannini EH, Ruperto N, Ravelli A, Lovell DJ, Felson DT, Martini A . Preliminary definition of improvement in juvenile arthritis. Arthritis Rheum 1997;40:1202–9.
Quartier P, Allantaz F, Cimaz R, et al. A multicentre, randomised, double-blind, placebo-controlled trial with the interleukin-1 receptor antagonist anakinra in patients with systemic-onset juvenile idiopathic arthritis (ANAJIS trial). Ann Rheum Dis 2011;70:747–54.
Ruperto N, Quartier P, Wulffraat N, et al.; Paediatric Rheumatology International Clinical Trials Organisation. A phase II, multicenter, open-label study evaluating dosing and preliminary safety and efficacy of canakinumab in systemic juvenile idiopathic arthritis with active systemic features. Arthritis Rheum 2012;64:557–67.
Ruperto N, Brunner HI, Quartier P, et al.; PRINTO; PRCSG. Two randomized trials of canakinumab in systemic juvenile idiopathic arthritis. N Engl J Med 2012;367:2396–406.
Goldbach-Mansky R, Shroff SD, Wilson M, et al. A pilot study to evaluate the safety and efficacy of the long-acting interleukin-1 inhibitor rilonacept (interleukin-1 Trap) in patients with familial cold autoinflammatory syndrome. Arthritis Rheum 2008;58:2432–42.
Hoffman HM, Throne ML, Amar NJ, et al. Efficacy and safety of rilonacept (interleukin-1 Trap) in patients with cryopyrin-associated periodic syndromes: results from two sequential placebo-controlled studies. Arthritis Rheum 2008;58:2443–52.
Hashkes PJ, Spalding SJ, Giannini EH, et al. Rilonacept for colchicine-resistant or -intolerant familial Mediterranean fever: a randomized trial. Ann Intern Med 2012;157:533–41.
Petryna O, Cush JJ, Efthimiou P . IL-1 Trap rilonacept in refractory adult onset Still’s disease. Ann Rheum Dis 2012;71:2056–7.
Lovell DJ, Glass D, Ranz J, et al. A randomized controlled trial of calcium supplementation to increase bone mineral density in children with juvenile rheumatoid arthritis. Arthritis Rheum 2006;54:2235–42.
Yokota S, Miyamae T, Imagawa T, et al. Therapeutic efficacy of humanized recombinant anti-interleukin-6 receptor antibody in children with systemic-onset juvenile idiopathic arthritis. Arthritis Rheum 2005;52:818–25.
Woo P, Wilkinson N, Prieur AM, et al. Open label phase II trial of single, ascending doses of MRA in Caucasian children with severe systemic juvenile idiopathic arthritis: proof of principle of the efficacy of IL-6 receptor blockade in this type of arthritis and demonstration of prolonged clinical improvement. Arthritis Res Ther 2005;7:R1281–8.
Yokota S, Imagawa T, Mori M, et al. Efficacy and safety of tocilizumab in patients with systemic-onset juvenile idiopathic arthritis: a randomised, double-blind, placebo-controlled, withdrawal phase III trial. Lancet 2008;371:998–1006.
De Benedetti F, Brunner HI, Ruperto N, et al.; PRINTO; PRCSG. Randomized trial of tocilizumab in systemic juvenile idiopathic arthritis. N Engl J Med 2012;367:2385–95.
DeWitt EM, Kimura Y, Beukelman T, et al.; Juvenile Idiopathic Arthritis Disease-specific Research Committee of Childhood Arthritis Rheumatology and Research Alliance. Consensus treatment plans for new-onset systemic juvenile idiopathic arthritis. Arthritis Care Res (Hoboken) 2012;64:1001–10.
Ruperto N, Lovell DJ, Quartier P, et al.; Paediatric Rheumatology International Trials Organization and the Pediatric Rheumatology Collaborative Study Group. Long-term safety and efficacy of abatacept in children with juvenile idiopathic arthritis. Arthritis Rheum 2010;62:1792–802.
Record JL, Beukelman T, Cron RQ . Combination therapy of abatacept and anakinra in children with refractory systemic juvenile idiopathic arthritis: a retrospective case series. J Rheumatol 2011;38:180–1.
Wulffraat NM, Brinkman D, Ferster A, et al. Long-term follow-up of autologous stem cell transplantation for refractory juvenile idiopathic arthritis. Bone Marrow Transplant 2003;32:Suppl 1:S61–4.
Brinkman DM, de Kleer IM, ten Cate R, et al. Autologous stem cell transplantation in children with severe progressive systemic or polyarticular juvenile idiopathic arthritis: long-term follow-up of a prospective clinical trial. Arthritis Rheum 2007;56:2410–21.
Abinun M, Flood TJ, Cant AJ, et al. Autologous T cell depleted haematopoietic stem cell transplantation in children with severe juvenile idiopathic arthritis in the UK (2000-2007). Mol Immunol 2009;47:46–51.
Author information
Authors and Affiliations
Corresponding author
Supplementary information
Supplementary Appendix
(DOC 30 kb)
Rights and permissions
About this article
Cite this article
Correll, C., Binstadt, B. Advances in the pathogenesis and treatment of systemic juvenile idiopathic arthritis. Pediatr Res 75, 176–183 (2014). https://doi.org/10.1038/pr.2013.187
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/pr.2013.187
This article is cited by
-
Systemic juvenile idiopathic arthritis: frequency and long-term outcome in Western Australia
Rheumatology International (2023)
-
Morbus Still – Ähnlichkeiten und Differenzen zwischen juveniler und adulter Form
Zeitschrift für Rheumatologie (2022)
-
S100A8/A9 is not essential for the development of inflammation and joint pathology in interleukin-1 receptor antagonist knockout mice
Arthritis Research & Therapy (2021)
-
Juvenile idiopathic arthritis: from aetiopathogenesis to therapeutic approaches
Pediatric Rheumatology (2021)
-
Extreme thrombocytosis in systemic juvenile idiopathic arthritis. A case report
Italian Journal of Pediatrics (2019)
