
Abstract
Invasive assays and lung tumor-bearing mice models using a human lung adenocarcinoma cell line A549 cells transfected with the Klotho (KL) gene, A549/KL cells, have confirmed that KL suppresses invasive/metastatic potential. This study aimed to identify the co-expression protein networks and proteomic profiles associated with A549/KL cells to understand how Klotho protein expression affects molecular networks associated with lung carcinoma malignancy. A two-step application of a weighted network correlation analysis to the cells’ quantitative proteome datasets of a total of 6,994 proteins, identified by mass spectrometry-based proteomic analysis with data-independent acquisition (DIA), identified one network module as most significantly associated with the A549/KL trait. Upstream analyses, confirmed by western blot, implicated the pro-apoptotic Bim (Bcl-2-like protein 11) as a master regulator of molecular networks affected by Klotho. GeneMANIA interaction networks and quantitative proteome data implicated that Klotho interacts with two signaling axes: negatively with the Wnt/β-catenin axis, and positively by activating Bim. Our findings might contribute to the development of future therapeutic strategies.
Similar content being viewed by others


Introduction
Lung cancer is the most common cancer worldwide, accounting for 2.5 million new cases and 1.5 million deaths in 2019, among which non-small-cell lung carcinoma predominantly accounts for more than 80%1. Even though many patients receive early diagnoses with low-dose spiral computed tomography, the 5-year survival rate remains less than 20% among patients who received chemo-, targeted, and immunotherapies. These deaths are thought to be because either those patients were diagnosed at an advanced stage or they received an early diagnosis that was incorrect1.
The klotho (KL) gene encodes a type-I membrane protein related to beta-glucuronidases and has three subfamilies: α-Klotho, β-Klotho (KLB), and γ-Klotho. KL refers to α-Klotho, which was first identified in a study of KL-deficient mice that developed multiple premature aging syndromes, contrasted with KL-overexpression associated with the extended lifespan of the mice2,3. Thus, KL received great attention as a new anti-aging gene that critically regulates aging and the development of age-related diseases. On the other hand, the KL gene has been recently highlighted in its association with tumor growth and invasion in various cancers, including breast, pancreatic, ovarian, lung, colon, and melanoma4. It is known that KL is involved in various biological processes and inhibits the insulin-like growth factor (IGF-1) signaling pathway5, which may be associated with malignancies of non-small-cell lung carcinoma (NSCLC) and small-cell carcinoma (SCLC)6.
Recently, Zhou et al. found that the concentrations of serum KLB were considerably higher in patients with NSCLC than in the control group and that KLB expression was significantly increased in patients after chemotherapy and epidermal growth factor receptor tyrosine kinase inhibitor (EGFR-TKI) targeted therapy as well as showing good correlation with progression-free survival (PFS) and overall survival (OS)7. Chen et al. constructed human lung cancer A549 cell lines transfected with Klotho or Klotho-specific shRNAs to investigate overexpression or knockdown Klotho in vitro. They suggested that Klotho can inhibit proliferation and increase apoptosis of A549 cells, partly due to the inhibition of IGF-1/insulin pathways and involving regulation of BAX/BCL2 expression (apoptosis-related genes)8. The authors suggested that the KL gene can potentially suppress tumors.
As described previously, we established an A549/KL cell line with stable and high Klotho protein expression by transfecting green fluorescent protein (GFP)-klotho plasmids into lung adenocarcinoma A549 cells9. It has been demonstrated in the A549/KL cell line that highly expressed KL significantly suppressed N-cadherin (CDH2) expression, an endothelial mesenchymal transition (EndMT)-related protein, which is also a favorable prognostic factor in lung cancer10,11. This observation formed the basis of a hypothesis that the KL gene induces inhibition of metastasis and invasiveness of lung cancer cells. Clinically, we sought to learn how the KL gene and protein affect malignancies, to develop treatment strategies to improve outcomes of patients with lung cancer.
A critical need is to identify molecular networks induced by the KL gene and its upstream regulators, which might help elucidate the mechanisms underlying the suppression of cancer malignancies. Recent advances in mass spectrometry (MS) have made MS-based proteomics much more powerful for use in shotgun protein sequencing and quantitative analysis of proteins expressed in clinical specimens than in the past. Quantitative proteome data can be used to identify key disease-related proteins and therapeutic targets in oncology12. We have used a high-performance mass spectrometer operated in the data-independent acquisition (DIA) mode to perform label-free ion intensity-based quantitative proteomics. We used the universal automated software suite DIA-NN (DIA-neural networks), which is particularly useful for performing high-throughput proteomics by enabling fast and reliable protein identification13.
This study aimed to identify co-expression protein networks associated with A549/KL cells, compared with those of A549 cells, to understand how Klotho protein expression affects molecular networks associated with the malignant nature of lung carcinoma cells. Weighted gene co-expression network analysis (WGCNA), an unsupervised clustering method based on correlation network expression14,15 was applied to quantitative proteome datasets. Then, Ingenuity Pathway Analysis was used to perform upstream analysis16 of data-driven protein co-expression networks, and GeneMANIA17 was used to identify interaction networks of proteins expressed characteristically in the A549 and A549/KL cells, respectively.
Results
Invasion assay
A CytoSelect 24-well cell invasion assay kit was used to evaluate the invasive ability of the A549 and A549/KL cells. A fluorescence method was used to quantify the infiltrating cells, and the average values were compared. The results showed that the invasive capacity was considerably reduced for the A549/KL cells than for the parental A549 cells (Fig. 1A).

(A) Invasion ability of A549 and A549/KL cells determined using a cell invasion assay kit. Three samples of each cell were quantified. The mean number of invasive cells was compared, indicating a statistically significant reduction in invasive ability for the A549/KL groups (p = 0.036). (B) Representative extracted lungs of mice. (B-1) The A549 cell-injected mice developed metastases in their lungs with multiple nodules on the surface of the lungs (a), but the A549/KL mice never developed metastases in their lungs (b). The lung nodule is indicated with an arrow. (B-2) Representative images of hematoxylin and eosin (H&E) staining of the lung sections of the A549 group. Image b is an enlarged view of image a. (C) A549 and A549/KL cells (5.0 × 106 cells) were injected into the tail veins of mice (three mice/group) to generate lung tumor metastases. Fifteen sections of lung tissue from three mice, five sections of lung tissue per mouse, were evaluated. Comparison of the mean number of metastases per cross-section showing the reduced metastatic ability of the A549/KL groups with a statistical significance of p = 0.0217.
Metastasis model of tumor-bearing mice
A lung tumor-bearing mice model was used to determine if high KL gene expression can suppress lung metastasis. A549 or A549/KL cells were injected into the tail vein of three mice each. After 8 weeks, the lungs were removed and pathologically diagnosed (Fig. 1B). Metastatic tumors of A549 were observed macro- and microscopically, as shown in Fig. 1B-1 and B-2, respectively, whereas no metastasis was observed in the A549/KL cells. There was a significant difference in the frequency of lung metastasis between the A549 and A549/KL cells; no lung metastasis occurred in mice that received A549/KL cells, confirming that KL expression suppressed cancer cell metastasis (Fig. 1C).
MS-based proteome datasets of A549 and A549/KL cells
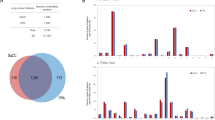
In-depth MS-based proteomic analysis was performed for the A549 adherent cells and A549/KL cells transfected with the KL gene (n = three samples each), resulting in a total of six samples. Each sample contained approximately 2 × 106 cells. A total of 6,994 proteins were identified, of which 6,958 (99.49%) were commonly expressed in the A549 and A549/KL cells, and only 36 (0.51%) proteins were unique to the A549/KL cells, a finding that showed an extremely high protein expression similarity between the cell lines (Fig. 2A). Volcano plots of the protein expression were generated using SimpliFi™ software (PROTIFI, Farmingdale, NY, USA; https://simplifi.protifi.com/) (Fig. 2B), exhibiting highly different expression levels. In the analysis, upregulated expressions of 215 and 302 proteins (p value < 0.05 and |log2 (Fold Change: [A549/KL]/[A549])|> 1) were shown for the A549/KL and A549 cells, respectively.

(A) Venn map of the proteins identified by mass spectrometry-based analysis and (B) their volcano plots, where proteins with p value < 0.05 and |log2 (Fold Change: [A549/KL]/[A549])|> 1 are indicated by red-filled circles. (C) Sample dendrogram and trait heatmap for the A549 (n = 3) and A549/KL (n = 3) with the triplicate measurements, which were constructed by the Euclidian distance-based network used in the weighted gene co-expression network analysis (WGCNA) software. (D) Pairwise Pearson correlations performed for the 60 identified modules regarding the connectivity measure (kME) of the module eigen-protein (correlation coefficient: Pearson’s r; heatmap order: eigenvectors; agglomeration method: complete; the number of clusters: 2).
Identification of co-expression protein networks by WGCNA
Following hierarchical clustering of the samples based on protein abundance (Fig. 2C), we used dynamic tree-cut, block-wise, and manual hybrid methods to perform a WGCNA analysis 14 utilizing the adjacency of an unsigned network with a soft threshold power of 10 (which was selected to approximate a scale-free topology), a minimum module size of 10, and a module detection sensitivity (deepSplit) of 4 (Figure S1). Then, we merged highly correlated modules to obtain the total protein cluster dendrogram. Correlations between the resultant modules and traits were obtained to identify protein modules that were significantly associated with the respective traits. Pairwise correlations between the sixty WGCNA modules identified were obtained regarding the connectivity measure, kME, of the module eigen-protein (Fig. 2D). A module is significant to a trait when its correlation r is > 0.5 and the q value is < 0.05.
A module-trait relationship obtained for sixty protein network modules (Fig. 3A) showed that 20 and 5 modules were significant to the A549 and A549/KL traits, respectively. We focus on modules significantly associated with the A549/KL trait. The WM55 (turquoise) module was found to have the highest significant association with the A549/KL trait (r = 1.0; q value = 1.01 × 10−17). However, the WM55 (turquoise) module has 2,150 module member proteins, which was too many to conduct further network analysis. Then, we decided to apply the second WGCNA analysis to those modules. Finally, the WM55-2 (turquoise-2) module consisting of 582 member proteins was identified as by far the most highly significant to the A549/KL trait (r = 0.97 and q value = 8.22 × 10−11) (Fig. 3B). The module membership of module proteins (kME vs. gene/protein significance) for the WM55-2 (turquoise-2) module showed a qualified correlation and a significant association with the A549/KL trait (Supplementary Figure S2).

Module-trait relationships. (A) the first step of the WGCNA analysis of 6,994 of proteins expressed. (B) the second step of the WGCNA analysis of the WM55 (turquoise) module (the number of proteins n = 2,150). Benjamini–Hochberg-corrected q-values were presented as a multiple statistical test.
Using the Search Tool for the Retrieval of Interacting Genes/Proteins (STRING) database version 11.5 (https://string-db.org/)18, the protein–protein interaction (PPI) networks of the WM55-2 (turquoise-2) module was reconstructed with Cytoscape (version 3.9.1) software (Institute for Systems Biology, Seattle, WA, USA: https://cytoscape.org/) (Figure S3). The cytoHubba plugin with maximal clique centrality was used to calculate the top ten hub proteins19. In this data-driven protein co-expression network, hub proteins are denoted in red-to-orange fill colors.
Upstream analysis by ingenuity pathway analysis (IPA)
Upstream analysis for the WM55-2 (turquoise-2) module was performed with IPA software (http://www.ingenuity.com)16. The top 20 master regulators (genes, RNAs, and proteins) together with participating regulators, canonical pathways, and regulator effects predicted for the turquoise-2 module are listed in Supplementary Table S1.
Top upstream regulators predicted to be activated (z > 2.0) include ERN1 (IRE1), MYC, EGFR, NFE2L2, XBP1, IL4, FN1, TGFB1, and CEBPB in the significance order of the overlap p-value. Top highly activated causal networks (z > 4.0) include CXCL14, BBC3 (BCL2 binding component 3, also known as JFY-1 or Puma), Rasgrp (RASGRP1-3), PRRG4, BCL2-like 11(BCL2L11, also known as apoptosis facilitator, Bim), RAS, and Egfr-Erbb2 in the significance order of the network bias-corrected p-value. Thus, those regulators might be categorized into carcinogenesis or pro-apoptotic functions. NFE2L2 is the master regulator of hypoxia and co-activation of ERN1(IRE1) and XBPI is associated not only with unfolding protein response (UPR) in ER stress (the inositol requiring enzyme 1-alpha (IRE1α)/X-box binding protein 1 pathway), but also with cell death and/or the apoptotic process20. Overexpression of fibronectin 1 (FN1) is associated with tumor progression by promoting proliferation, invasion, and metastasis21. Most of the top regulator effects predicted were annotated characteristically as apoptosis of carcinoma cell lines, including lung cancer (Table S1). Therein, it should be noted that only Bim and Puma involve apoptosis regulator BAX (BAX) and Bcl-2-related ovarian killer protein (BOK) as their participating regulators among the top causal networks (Table 1). Indeed, our proteomic analysis captured BAX and upregulated BOK in the A549/KL cells (Table 2), implicating that both Bim and Puma are the most likely candidates for a master regulator affected by the KL gene.
Canonical pathways predicted to be activated were the UPR, EIF2 signaling, NRF2-mediated oxidative stress response, insulin-secretion signaling pathway, pulmonary fibrosis idiopathy signaling pathway, regulation of the epithelial–mesenchymal transition (EMT) by growth factors pathway, and EGF signaling (Table S1).
The integrative causal networks of Bim and Puma (presented in Figure S4) constructed by IPA implicate that Bim and Puma are unphosphorylated by inactivated MEK–MAPK cascades downstream of fibroblast growth factor receptor 1 (FGFR1) and inhibit the apoptosis checkpoint molecule BCL2, being degraded via the proteasome. Then, inactivated BCL2L1/BCL-XL allows BOX, BAX, and Bcl-2 homologous antagonist/killer (BAK1) activation, responding to ER stress through the IRE1α-XBP1 UPR pathway. The activated BAX/BAK1 promotes the release of CYC1 (cytochrome c 1) from mitochondria, which would form apoptosomes together with CASP9 (caspase 9) and CASP3 (caspase 3).
GeneMANIA-based upstream analysis
Among the total proteins identified, we focused on the 23 adhesion- and apoptosis-related proteins upregulated between A549 and A549/KL (Table 2). GeneMANIA upstream analysis (University of Toronto, Ontario, Canada: http://genemania.org/)17 via Cytoscape app (version 3.9.1) (https://cytoscape.org/)18 was applied to those proteins to find an additional 20 interacting genes/proteins (Fig. 4). Proteins upregulated in the A549 or A549/KL cells are shown as large black-filled circles, and their upstream and/or interacting molecules that were found are shown as gray-filled circles, which are ordered by relatedness to the query genes/proteins in bipartite layouts (Fig. 4A)17.

(A) The GeneMANIA analysis of the 23 adhesion and apoptosis-related proteins unregulated in the (a) A549 and (b) A549/KL cells, each other. Red circles denote proteins associated with the Wnt signaling pathway, and pink circles are associated with the apoptosis pathway. (B) Top thirty annotations on GO biological processes obtained by GeneMANIA for the A549 and A549/KL cells (q-values denote the Benjamini–Hochberg FDR multiple testing correction).
Regarding the A549 cells, the 10 adhesion- and apoptosis-related proteins upregulated include CDH1 (E-cadherin), epithelial cell adhesion molecule (EpCAM), CTNNB1 (β-catenin), BCL-6 corepressor-like protein 1 (BCORL1), and carcinoembryonic antigen (CEACAM5) and were found to interact with catenin α-1, SRC, EGFR, MET, LEF1 (TCF1-α), and FGFR1. The biological process (GO) annotated to the molecular networks found that A549 cells included extrinsic components of membranes, cell–cell junction, cell–cell adhesion via plasma–membrane adhesion molecules, negative regulation of cell adhesion, and negative regulation of binding (Fig. 4B). Their related networks included E-cadherin signaling events, the Wnt signaling pathway, and pathways regulating HIPPO signaling. Characteristically, the key molecules of the Wint/β-catenin signaling pathway included β-catenin, catenin α-1, E-cadherin, TCF1-α, and TCF7L2 (T-cell factors/lymphoid enhancer factors), CTNNA3 (α-T-catenin), CTNNA2 (α-N-catenin), and CDH17 (Cadherin-17) (Fig. 4Aa). β-catenin has the leading role in the Wnt signaling pathway associated with EMT in cancer progression. Upregulation of N-cadherin (CDH2) followed by downregulation of E-cadherin, which is referred to as the “cadherin switch,” is the hallmark of EMT, by which β-catenin activates several key signaling pathways, such as Wnt/β-catenin and the lymphoid enhancer factor (LEF)/T-cell factor (TCF), leading to the acquisition of invasive and metastatic potential. Canonical Wnt signaling leads to the accumulation of β-catenin in the cell membrane, and β-catenin unphosphorylated is stabilized in the cytoplasm, followed by its translocation into the nucleus to activate the transcription of TCF/LEF-target genes, key factors in cell proliferation and invasion, including FN1 and c-Myc22,23. Cadherin-17 belongs to the Wnt/β-catenin signaling pathway, and it was shown that targeting the cadherin-17 gene (CDH17) by RNA interference-mediated knockdown inhibited the proliferation of both primary and highly metastatic hepatocellular-carcinoma (HCC) cell lines in vitro and in vivo24.

(A) Western blot analysis for A549 cells and A549/KL cells using anti-Klotho, anti-Bim, anti-Puma, anti-β-catenin, anti-Phospho-β-Catenin, and anti-Wnt5a antibodies. Wnt5a is a marker of non-canonical Wnt-Frizzled pathway. All gel images are presented in Figure S6 as full as possible length gels and blots with membrane edges visible. (B) The graph shows the relative expression levels of Bim and Puma in the A549 and A549/KL cells quantified by ImageQuant TL ver.8.1 (Cytiva) and demonstrates highly upregulated Bim levels in the A549/KL cells, whereas Puma was detected only at a background level.
Our proteomic analysis could not capture N-cadherin but could capture E-cadherin, as Table 2 and Fig. 4Aa show, suggesting that our A549 strain did not acquire a highly enhanced but rather a limited mesenchymal characteristic for invasion and/or metastasis. The extent of the “cadherin switch” seems to depend on a balance between unstable phosphorylated β-catenin and stable unphosphorylated β-catenin in the cytoplasm. The former leads to proteasomal degradation and the latter is translocated into the nucleus. N-cadherin binds to β-catenin, and the expression levels of N-cadherin seem to depend on the balance of β-catenin accumulated between the cell surface and the cytoplasm, where Wnt/β-catenin signaling functions as a rheostat23. Nevertheless, our GeneMANIA analysis results of upregulated proteins associated with the A549 strain (Fig. 4Aa) implicated the activation of canonical Wnt/β-catenin signaling. The main proteins participating in the Wint/β-catenin25 and the Wnt signaling pathways, together with matrix metalloproteinases, from our proteome data are listed in Table 2C and D.
On the other hand, the 13 adhesion- and apoptosis-related proteins upregulated in the A549/KL cells include BCAM, neuronal cell adhesion molecule (NRCAM), protocadherin 9 (PCDH9), protocadherin Fat1 (FAT1), BOK, Bcl-xL/Bcl-2–associated death promoter (BAD), and induced myeloid leukemia cell differentiation protein Mcl-1 (MCL1). Interestingly, their interacting molecules were apoptosis checkpoint molecules, including Bcl2-interacting mediator of cell death (BCL2L11, Bim), p53 upregulated modulator of apoptosis (BBC3, Puma), BAK1, BAX, BCL2, BCL2L1, B-cell lymphoma-extra large (Bcl-XL or BCL-XL), and BH3-interacting domain death agonist (BID). Other apoptosis-related molecules were FAS-associated death domain protein (FADD), apoptosis-mediating surface antigen FAS (FASLG), apoptotic protease activating factor-1 (APAF1), PMA-induced protein 1 (PMAIP1), AKT1, CASP3, CASP7, CASP8, and CASP9 (Fig. 4Ab).
The results obtained above are consistent with those from IPA-based upstream and causal network analysis, supporting that both Bim and Puma are most likely master regulators of molecular networks affected by the KL gene. Finally, our western blot analysis confirmed Bim as a possible master regulator in molecular networks altered by the KL gene transfection to the A549 cells (Fig. 5).
A web-based survival analysis (KMplot) for mRNA data of lung carcinoma indicated that better overall survival was associated with the upregulated levels of Bim, BOK, and MCL1 than with BLC2L1 (BCL2-XL) and N-cadherin (Figure S5)26.
Several key molecules relevant to the Wnt signaling pathway were also found, which include PCDH9 and FAT1. PCDH9 inhibits EMT and cell migration27, and FAT1 is known as a tumor suppressor, whereas it is frequently mutated, and inhibits Wnt/β-catenin signaling by negatively regulating β-catenin nuclear translocation and its transcriptional activity28,29. The biological processes (GO) dominantly annotated were the extrinsic and intrinsic apoptosis signaling pathway, apoptotic mitochondrial changes, positive regulation of mitochondrial membrane permeability involved in the apoptotic process, the release of cytochrome c from mitochondria, and positive regulation of mitochondrial outer membrane permeabilization involved in the apoptotic signaling pathway (Fig. 4B).
Activation of Bim was reported to suppress cancer metastasis by inhibiting MCL130,31 and EMT-related N-cadherin expression11. Conversely, it has been shown that upregulation of N-cadherin suppresses Bim expression32. Our study has not provided evidence that Klotho directly interacts with N-cadherin or directly perturbs the Wnt/β-catenin signaling and apoptosis signaling via Bim. However, it was suggested that the extracellular domain of Klotho binds to several Wnt ligands, which inhibit the potential ability to activate Wnt signaling33. Klotho is the co-receptor of FGFR1, which is the specific receptor for the phosphaturic hormone fibroblast growth factor-23 (FGF23). There are two types of Klotho proteins: soluble Klotho (sKlotho) and secreted Klotho. The sKlotho protein binds to multiple ligands of Wnt and suppresses various gene transcriptions. Upregulated sKlotho has previously been demonstrated to attenuate renal fibrosis by suppressing Wnt signaling in mice models34,35. Accumulating data have suggested crosstalk between Wnt/β-catenin signaling and regulation of Klotho and FGF2336.
In this study, the Klotho protein and various key proteins associated with cancer-, apoptosis-related, and Wnt/β-catenin pathways were quantitively identified (Table 2). A high expression level of the Klotho protein was observed in the A549/KL cells, confirming its successful establishment of the A549/KL strain, whereas the detected upregulation of the EMT markers VIM and ZEB1 might reflect a complicated trait of the A549/KL strain, but this remains unclear.
Our MS-based proteomic analysis captured several apoptosis-related proteins, including BAK1, BAX, Bax inhibitor 1 (BI1), BCL2L1 (BCL-XL), APAF1, FADD, and death receptors DR4/5. Upregulation of BAK1 and downregulation of BAX, BI1, and BCL-XL were observed in A549/KL. Upregulated expression of the death receptor 4 TNF-related apoptosis-inducing ligand-receptor 1 (TRAILR1) and death receptor 5 (DR5) TRAILR2 was observed in A549/KL, which might be attributed to release from the selective inhibition by EMT37.
Regarding the A549/KL cells, we identified the co-receptor of Wnt ligands, slightly upregulated Frizzled-2/5/6/7 and low-density lipoprotein receptor-related protein 5/6 (LRP5/6), and slightly upregulated GSK3β. Those proteins form the signalosome complex, in which LRP5/6 are phosphorylated when the Wnt signaling is activated (Wnt ON). Proto-oncogene Src directly phosphorylates LPR6 and Frizzled, which was observed to be downregulated in A549/KL. Notably, the phosphorylation of a protein/peptide generally reduces its electrospray ionization efficiency in MS. The above observations likely reflect inactivated Wnt signaling (Wnt OFF) in A549/KL. Wnts were not detected, but Wnt ligand secretion mediator, also known as Wntless (WLS), which is required for the secretion of all Wnts, was detected in both traits38.
Moreover, upregulation of fibroblast growth factor 2 (FGF2), FGFR1, FGFR4, and EGFR were also found in A549/KL (Table 2B). The binding of FGF2 to FGFR1, the most important FGF2 receptor, activates downstream signaling, including MAPK/ERK, PLCg, and PI3K/AKT pathways39. Upregulation of FGFR1 and FGF2 in the A549/KL cells might suggest that FGF2-activated FGFR1 signaling induces FGFR1 internalization, thereby stabilizing FGFR1 expression40. Upregulation of FGFR1 in NSCLC cell lines by hypoxia induces subsequent activation of MAPK cascades, leading to attenuated induction of the pro-apoptotic factor Bim, which drives acquired resistance to EGFR tyrosine kinase inhibitors41,42,43,44. Therefore, combining EGFR TKIs with FGFR1 inhibitors or MEK inhibitors is considered an attractive therapeutic strategy for NSCLCs41.
Discussion
The results of our invasive assay and lung tumor-bearing mice model confirmed that Klotho suppresses invasive and metastatic potential, in which N-cadherin suppression is assumed to be induced by the KL gene11. We conducted in-depth MS-based quantitative proteomic analysis to elucidate protein expression profiles of A549 and A549/KL cells. The proteomic data successfully applied to WGCNA analysis identified 60 data-driven WGCNA co-expression modules associated with the A549 and A549/KL cells. Additionally, the two-step WGCNA analysis of the obtained MS-based proteomic data identified one WGCNA module, the WM55-2 (turquoise-2) consisting of 582 member proteins as most significantly associated, by far, with the A549/KL cells. Both the upstream regulator and causal network analysis by IPA for the turquoise-2 module and GeneMANIA analysis applied to the adhesion- and apoptosis-related proteins upregulated in the A549/KL cells implicated Bim and/or Puma as the most likely master regulators underlying molecular networks affected by Klotho occurring in the A549/KL cells. Our western blotting analysis confirmed Bim as a significant regulator (Fig. 5).
Involvement of canonical Wnt signaling is most likely responsible for the invasive and metastatic potential of the A549 trait, and was inactivated in the A549/KL cells, which might be evidenced by the expressions of PCDH9 and Fat1. Loss of N-cadherin could be explained partly by the “cadherin switch” regulated centrally by β-catenin in the context of inactivated Wnt signaling. The GeneMANIA analysis results implicated the involvement of two different signaling axes: FGF-FGFR and Wnt/β-catenin. Interestingly, our quantitative proteomic data exhibited upregulated expression of the FGFR1 protein in A549/KL cells. A speculative scenario is that Klotho suppresses Wnt/β-catenin signaling and abolishes phosphorylation downstream of MEK/ERK in the FGF-FGFR signaling, which prevents phosphorylation of Bim and synergically Wnt co-receptor LRP640,44.
Klotho appears to function as an antagonist of the Wnt signaling pathway. It can inhibit the activation of this pathway. The canonical Wnt pathway involves the stabilization and nuclear translocation of β-catenin, which leads to the expression of specific target genes associated with cell proliferation and survival. Klotho, especially the secreted form known as sKL, has been shown to reduce the active form of β-catenin (non-phosphorylated or dephosphorylated on specific residues) and decrease the expression of Wnt target genes such as c-Myc and Cyclin D145. It was also suggested that Klotho acts as a tumor suppressor and an inhibitor of the Wnt/β-catenin pathway in HCC46. A simple diagram summarizing the results of this study together with the literature regarding Klotho, BIM, and Wnt/β-catenin signaling47,48,49,50 is presented in Fig. 6.

A model of the cross-talk between FGF-FGFR and Wnt/β-catenin signaling in the A549 cells, with which Klotho negatively interacts in the A549/KL cells. When Wnt/β-catenin signaling is activated (Wnt ON), Wnts bind to the Wnt coreceptors Frizzled receptors (FZDs) and LRP5/6, followed by phosphorylation of LRP5/6, and induce the clustering of Wnt/Frizzled/LRP6 into the signalosome complexes, which allows for Axin and GSK3β binding to them. The signalosomes amplify the Wnt signal, where phosphorylated LRP5/6 triggers the successive recruitment of Axin1/GSK3β/β-catenin to the cytoplasmic tail of LRP5/6, resulting in the displacement of GSK3β from the Axin1/β-catenin complex, inhibition of GSK3β and decreased phosphorylation of β-catenin. Thus, β-catenin is stabilized and accumulated as free β-catenin in the cytosol, which is subsequently translocated into the nucleus and activates the TCF (T-cell factor) /LEF (lymphoid enhancer factor)-dependent transcription of Wnt target genes. N-cadherins are linked to the actin cytoskeleton participating in the adherens junction, through their binding to α-catenins and β-catenins. The release of β-catenin from adherens junctions is induced by oscillatory fluid flow, and Wnt/β-catenin signaling competes for the same cellular pool of β-catenin. Activated FGF-FGFR signaling turns on downstream signaling such as the RAF–MEK–ERK and PI3K–PKB pathways. Both pathways promote cell survival by antagonizing pro-death proteins, Bim and BAD. ERK directly phosphorylates Bim, followed by its proteasomal degradation47, and also PKB can directly phosphorylate BAD48. Both the ERK and PKB also promote the expression of pro-survival proteins BCL-XL and MCL149,50. Klotho interacts negatively with the Wnt/β-catenin signaling (Wnt OFF), which results in the phosphorylation of cytoplasmic β-catenins followed by their rapid degradation, mediated by the destruction complex, and also induces Protocadherin 9 and Fat1, suppressing EMT and Wnt/β-catenin signaling by negatively regulating β-catenin nuclear translocation and its transcriptional activity. Klotho suppresses the downstream of FGFR signaling, ERK, and PKB, by which activated Bim and BAD promote pro-apoptosis signalings, through activated BAX/BAK, APAF1, and Caspases, and synergically which prevents phosphorylation of Wnt co-receptor LRP6. Moreover, Klotho induces activation of DR4/5 death receptor signaling.
A limitation of this study is the small sample size using only the A549 cell line. Further study is required to validate our results and identify which molecules targeted by Klotho are responsible for the A549/KL trait.
In conclusion, we successfully applied in-depth MS-based proteomics to identify and quantify proteins expressed in A549 and A549/KL cells. Upstream analyses using both WGCNA and GeneMANIA were successfully applied to their proteomic datasets and identified the functional protein networks significantly associated with both cell traits.
Materials and methods
Cell culture and transfection
A549, a human lung adenocarcinoma cell line was maintained in Dulbecco’s modified Eagle’s medium (DMEM; cat. no. D6429; Millipore Sigma) supplemented with 0.1% sodium bicarbonate, L-glutamine, sodium pyruvate, 10% heat-inactivated fetal bovine serum (FBS; cat. no. F2442; Millipore Sigma), and penicillin (100 U/mL) in a humidified atmosphere of 5% CO2 at 37 °C. The Lipofectamine 3000® transfection reagent (Invitrogen; Thermo Fisher Scientific Inc.) was used to transfect GFP–klotho plasmids A549 cells according to the manufacturer’s instructions. The transfection reagent was used to transfect 0.25 –1 × 106 cells with 2 µg of plasmid DNA, which were then incubated for 2 h in a humidified atmosphere of 5% CO2 at 37 °C. The cells were washed, and then medium that included FBS was added. The GFP–Klotho plasmid, which was previously provided by Dr. Nabeshima (Foundation for Biomedical Research and Innovation, Kobe, Japan), was transfected into the A549 cells, and 24 h later the cells were pelleted (almost 1 mg) by centrifugation at 1,500 rpm for 10 min and resuspended in phosphate-buffered saline (PBS) to a final density of ~ 2.9 × 106 cells/mL, and the suspension was then filtered through Cell Strainer (40 μm) to remove cell aggregates. A FACSCanto II (BD Biosciences) with the activation set at 488 nm and fluorescence-emission monitoring at 508 nm (GFP), was used to sort the GFP-positive cells. FlowJoTM v.10.7 software (TreeStar Inc.) was used to perform the data acquisition and analysis. At least 10,000 events were collected for each analysis. The forward-scatter and side-scatter parameters were used to eliminate the dead cells and debris, and the remaining cells were sorted into GFP-positive and GFP-negative populations 11.
Isolation of clones expressing Klotho
Following GFP–Klotho transfection, the GFP-positive cells were cultured with 0.1% sodium bicarbonate, L-glutamine, sodium pyruvate, and 10% FBS, penicillin (100 U/mL) in a humidified atmosphere of 5% CO2 at 37 °C for 2–3 weeks, and the colonies were then harvested. A limiting dilution method was used to obtain a single clone. Western blotting was performed to check several colonies for Klotho expression, and stably overexpressing Klotho cell line, A549/KL, was established11.
Cell invasion assay
A CytoSelect 24-well cell invasion assay kit (Cell BioLabs, Inc.) was used to perform cell invasion assays.This assay kit contains polycarbonate membrane inserts (8 μm pore size). The upper surface of the insert membrane is coated with a uniform layer of dried basement membrane matrix solution. The basement membrane layer serves as a barrier to discriminate invasive cells from non-invasive cells. Invasive cells can degrade the matrix proteins in the layer and ultimately pass through the pores of the polycarbonate membrane. Finally, these cells are dissociated from the membrane and subsequently detected by CyQuant® GR Dye (Invitrogen).
A cell suspension was placed in an upper chamber in serum-free media. A549 and A549/KL cell suspensions containing 3.0 × 105 cells were seeded in the chamber, and each cell was seeded at three different locations. After incubating for 24 − 48 h at 37 °C in a 5% CO2 atmosphere, the invasive cells were dissociated from the membrane by adding Cell Detachment Buffer to the lower chamber. Invasive cells were lysed by adding Lysis Buffer with CyQuant GR Fluorescent Dye. Subsequently, each sample was transferred to a 96‐well microtiter plate and quantified by reading the fluorescence at 480 nm/520 nm with a plate reader.
Mouse lung metastasis model
Female 5-week-old BALB/cSlc-nu/nu mice were obtained from MediRidge Company, Limited (Tokyo, Japan). All animal experiments were conducted according to protocols approved by the Animal Care and Use Committee of Nippon Medical School (approval number: 2021–030). A549 and A549/KL cells (5.0 × 106 cells) in 100 μL PBS were injected into the tail veins of mice (three mice/group) to generate lung tumor metastases. The mice injected with cells were sacrificed at 8 weeks. Excised mouse lungs were fixed in formalin and embedded in paraffin. Tumor metastasis to the lungs was assessed by hematoxylin and eosin (H&E) staining. Fifteen sections of lung tissue from three mice, five sections of lung tissue per mouse, were evaluated.
Sample preparation for LC–MS/MS
Reagents and materials
The protease inhibitor (Protease Inhibitor Cocktail Tablets, complete, Mini, EDTA-free Tablets) was purchased from Roche Diagnostics (Indianapolis, IN, USA). Benzonase (Benzonase® endonuclease, purity grade I (≥ 99%) suitable for biopharmaceutical production) was purchased from Merck (Whitehouse Station, NJ, USA). Ammonium bicarbonate (AMBIC), PBS, dithiothreitol (DTT), iodoacetamide (IAA), and triethylammonium bicarbonate (TEAB) were purchased from Sigma (St. Louis, MO, USA). Sequencing-grade trypsin was purchased from Promega (Madison, WI, USA). BCA reagent was purchased from Thermo Fisher Scientific (Pierce Biotechnology, Rockford IL, USA). Acetonitrile, trifluoroacetic acid, formic acid, and methanol were purchased from Kanto Chemical Co. Ltd. (Tokyo, Japan). Sodium dodecyl sulfate (SDS) was purchased from Amersham Biosciences (Amersham, UK). Phosphoric acid was purchased from NACALAI TESQUE, INC. (Kyoto, Japan). All water used in this study was Milli-Q ultrapure water (Merck Millipore, Billerica, Massachusetts, USA). All reagents were of analytical grade.
Sample preparations
The samples analyzed in this experiment consisted of A549 adherent cells and A549 cells transfected with the KL gene. For cell culturing, 75 cm2 culture flasks were used with w DMEM + 10% FBS medium added. Cells at 80% confluency were collected by the following procedure. First, the old medium was removed, and the cells were washed with 10 mL of PBS (–). Then, 2 mL PBS (–) was added, and the cells were scraped off with a cell scraper. The cell suspension was centrifuged at 1500 rpm for 3 min, and the pellet was resuspended in 1 mL PBS (–) and transferred to a 1.5 mL tube. The tube was centrifuged at 700 g for 10 min, and the collected cell pellet was stored at − 80 °C.
A 50-µL aliquot of lysis buffer was added to the harvested cell pellet sample. The component of the lysis buffer was as follows: protease inhibitor (prepared at a 14-fold dilution) and benzonase were added to 10% SDS in 10 mM TEAB, pH 7.55. A Pierce™ BCA Protein Assay Kit (Thermo) was used to quantify the dissolved protein sample solution to determine the protein concentration. All samples were prepared to have a total starting material of 100 μg protein.
The samples, A549 adherent cells, and A549 cells transfected with the KL gene were divided into three portions, resulting in six samples that were subjected to sample preparation using S-trap™51,52,53,54,55. For each of the six samples, DTT and IAA were used for reduction and alkylation, followed by bringing the samples back to room temperature and adding phosphoric acid to a final concentration of 1.2%. Then, S-trap buffer (100 mM TEAB in 90% methanol, pH 7.1) was added in a volume sixfold higher than that of the sample, and the mixture was applied to the S-trap micro spin column. The protein retained on the S-trap was washed five times with S-trap buffer. Then, a 1-μg equivalent of trypsin was added, and the samples were incubated at 47 °C for 1 h, then incubated overnight at 37 °C.
The samples were then eluted using the following method. First, 40 μL of 50 mM TEAB was added to the S-trap micro and centrifuged at 4000 g for 1 min. Then, 40 μL of 0.2% formic acid was added to the S-trap micro and centrifuged at 4,000 g for 1 min. Finally, 35 μL of 0.2% formic acid in 50% acetonitrile was added to the S-trap micro and centrifuged at 4,000 g for 1 min. The entire flow-through was collected, and the solvent was completely evaporated on a SpeedVac evaporator. The sample was then reconstituted in 20 μL of 0.1% trifluoroacetic acid in 2% acetonitrile for LC–MS/MS analysis.
Proteomic analysis by liquid chromatography–tandem mass spectrometry (LC–MS/MS)
The eluted samples were separated by nanoflow reversed‐phase LC followed by analysis on a Q-Exactive mass spectrometer (Thermo Fisher Scientific, San Jose, CA) equipped with a Dream spray nano-electrospray ionization source (Dream spray, AMR Inc., Tokyo, Japan). The LC instrument was an Ultimate 3000 dual‐solvent delivery system (Thermo Fisher Scientific) equipped with a PAL LSI auto‐sampler (CTC Analytics AG, Zwingen, Switzerland). The samples were loaded onto a capillary reversed-phase separation column packed with 1.6-μm-diameter gel particles with a 120 Å pore size (AURORA C18, 250 × 0.075 mm, IonOpticks). Eluent A was 0.1% formic acid, and eluent B was 100% acetonitrile. The column was eluted at a flow rate of 0.2 μL/min with a concentration gradient of A + 5% B to 35% B over 100 min and from 35% B to 95% B over 1 min, with subsequent isocratic elution at 95% B for 8 min and then a return to initial conditions from 95% B to 5% B over 1 min for re-equilibration.
The mass spectrometer was operated in DIA mode in which the MS acquisition with a mass range of m/z 380–1600 was automatically switched to MS2 acquisition under the automated control of Xcalibur software 3.1 (Thermo Fisher Scientific). DIA was performed with staggered isolation windows, a loop count of 25.0 m/z, and a normalized collision energy of 27 with a 200-ms maximum injection time at 70,000 resolution. In the DIA mode, each cycle consisted of an MS1 scan of 380–1600 m/z with 70,000 resolution and an AGC target of 1 × 106, followed by 48 MS2 scans of 400–1600 m/z with a resolution of 35,000 and an AGC target of 1 × 106 56.
Protein identification
Acquired raw data were processed by DIA by Neural Networks (DIA-NN) for proteomics analysis. DIA-NN is known to be particularly useful for high-throughput proteomics applications because it improves the performance of protein identification and quantification in traditional DIA mode proteomics applications, enabling fast and reliable protein identification13,57.
The algorithm for DIA-NN is described as follows. DIA-NN version 1.8.1 in library-free mode was used with the same Uniprot FASTA database. The Protein Database was applied to the human database (UniProt Reference Proteome—Homo sapiens, Taxonomy 9606—Proteome ID UP000005640 -. 20,373 entries—UniProt release 2022_03, reviewed human canonical)13.
Precursors of charge state 1–4, peptide lengths 7–30, and peptide m/z-values from 300 to 1800 were considered with a maximum of one missed cleavage. A maximum of one variable modification per peptide was considered. We used cysteine carbamidomethylation as a fixed modification, N-terminal methionine excision as a variable modification, methionine oxidation as a variable modification, and N-terminal acetylation as a variable modification. Precursor False Discovery Rate (FDR) was then filtered at 1%13,57.
Weighted correlation network analysis (WGCNA)
The similarity in protein expression patterns for all protein pairs was calculated according to their pairwise Pearson’s correlation coefficient r (i.e., the similarity between proteins i and j was defined as (1–ri,j)/2, where ri,j is the Pearson’s correlation coefficient of the protein expression pattern between the two proteins). We performed a network topology analysis using the adjacency of an unsigned network, aij =|cor(xi, xj)|β between gene expressions x(i) and x(j), for various soft-thresholding powers ranging from β = 1 to 20 to choose an optimal value of balance between independence and mean connectivity, where A topological overlap matrix (TOM) that considers topological similarities between a pair of proteins in the network was then generated from the resultant scale-free co-expression network. We used dissimilarity according to TOM (1 − TOM) to generate a tree by hierarchical clustering, and dynamic tree-cutting to trim the branches to determine protein modules16.
The modules were summarized by the first principal component, which is referred to as eigen proteins in the text because they express the highest connectivity in the module. Module membership, defined as the correlation between the protein expression profile and the module eigen-protein, was measured with values ranging from 0 to 1, with “0” representing a gene that is not part of the module and “1” representing high connectivity with the module. Subsequently, the module-trait association was determined by the correlation between the module eigen-protein and the traits A549 and A549/KL. A protein module was summarized by the top hub protein (referred to as the “eigen-protein”) with the highest connectivity in the module. The two-step WGCNA analyses were performed using the WGCNA R-package14 implemented in RStudio.
Protein–protein interaction (PPI) network construction
We used the STRING database (version 11.5) to construct a PPI network for a protein module (https://string-db.org/)18. STRING networks were calculated under the criteria for linkage with experiments, databases, text mining, and co-expression using the default settings (medium confidence score: 0.400; network depth: 0 interactions). Functional enrichment results were obtained for canonical pathways with a p-value < 0.05. Protein networks were subsequently exported to Cytoscape (version 3.9.1) (https://cytoscape.org/) from the STRING database18. The hub proteins in each module were identified according to their intramodular connectivity and their correlation with module eigenproteins. The proteins inside the co-expression modules exhibit high connectivity and the proteins within the same module may have similar roles. The top 10 high-degree proteins were identified using the cytoHubba plugin19. The top-ranked proteins in each module were considered hub proteins and designated “highly connected proteins.” Functional enrichment results were obtained for canonical pathways by considering a network bias-corrected p value of < 0.05 for statistical significance.
Quantile normalization of protein expression data obtained by MS-based proteomic analysis conducted in DIA mode and the pairwise correlation of identified WGCNA modules were performed in JMP software (SAS Institute, Cary, NC, USA). The Intervene Shiny App was used to visualize pairwise correlation (https://intervene.shinyapps.io/intervene/)58.
Ingenuity pathway analysis (IPA)
IPA software was used to predict upstream regulators, causal networks, and canonical pathways16. Quantile-normalized protein expression data of the selected modules were used as input datasets. Both the upstream regulators and causal networks (p < 0.05) predicted from the WGCNA network modules were significantly associated with the A549 or A549/KL trait in which the activation and inhibition of a predicted network were defined by z-values that were > 2.0 and < − 2.0, respectively. The upregulation was defined by z-values > 1.5 and < 2.0, whereas downregulation was defined by z-values > − 2.0 and < − 1.5.
GeneMANIA
The list of proteins in gene name/accession number was submitted to the GeneMANIA (http://genemania.org/)17 via Cytoscape app (version 3.9.1) (https://cytoscape.org/), where an additional 20 related genes/proteins were searched with all the interaction networks consisting of co-expression, co-localization, genetic interactions, pathways, physical interactions, predicted, and shared protein domains with 20 attributes using GO biological-based weighting. Large black-filled circles are query genes/proteins, and their upstream and/or interacting molecules were shown in gray-filled circles, which are ordered by relatedness to the ten query genes/proteins in bipartite layouts. A colored relationship line corresponds to the respective interaction network category.
Western blot
Overnight incubation was performed at 4 ℃ with rabbit primary antibodies against BIM (#2933, 1:1,000; Cell Signaling Technology) and PUMA (#4976; 1:1,000; Cell Signaling Technology). After washing with Tris-buffered saline and 0.1% polysorbate 20, membranes were incubated with horseradish peroxidase–conjugated secondary anti-rabbit IgG antibodies (#7074; 1:1,000; Cell Signaling Technology) at room temp for 1 h. 1 h incubation was performed at RT with rabbit primary antibodies against β-catenin (#8480, 1:1,000; Cell Signaling Technology), Phospho-β-Catenin (Ser675) (#4176, 1:1,000; Cell Signaling Technology), Wnt5a (ab235966, 1:1,000; Abcam). Regarding Klotho-GFP, 1 h incubation was performed at RT with Rat primary antibodies against Klotho (KO603, 1:1,000; Medicinal Chemistry Pharmaceutical Co.) and horseradish peroxidase–conjugated secondary anti-Rat IgG antibodies (5220–0365; 1:20,000; SeraCare). Immunoreactive protein was detected with enhanced chemiluminescence substrate, and band intensities were quantified by ImageQuant TL ver.8.1 (Cytiva). After visualization of the target protein, membranes were stripped and reincubated with antibodies against β-actin (#4967; Cell Signaling Technology)11.
Data availability
The unfiltered MS datasets generated and analyzed in this study have been deposited in ProteomeXchange (http://proteomecentral.proteomexchange.org) and jPOST, with the dataset identifiers PXD042978 and JPST002200, respectively.
References
Siegel, R. L., Miller, K. D. & Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 69, 7–34 (2019).
Kuro-o, M. et al. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature 390, 41–51 (1997).
Kurosu, H. et al. Suppression of aging in mice by the hormone Klotho. Science 309, 1829–1833 (2005).
Rubinek, T. & Wolf, I. The role of alpha-klotho as a universal tumor suppressor. Vitam. Horm. 101, 197–214 (2016).
Ma, Z., Dong, A., Kong, M. & Qian, J. Silencing of the type 1 insulin-like growth factor receptor increases the sensitivity to apoptosis and inhibits invasion in human lung adenocarcinoma A549 cells. Cell. Mol. Biol. Lett. 12, 556–572 (2007).
Mattarocci, S. et al. Intracellular presence of insulin and its phosphorylated receptor in non-small cell lung cancer. J. Cell. Physiol. 221, 766–770 (2009).
Zhou, J., Ben, S., Xu, T., Xu, L. & Yao, X. Serum β-klotho is a potential biomarker in the prediction of clinical outcomes among patients with NSCLC. J. Thorac Dis. 13, 3137–3150 (2021).
Chen, B., Wang, X., Zhao, W. & Wu, J. Klotho inhibits growth and promotes apoptosis in human lung cancer cell line A549. J. Exp. Clin. Cancer Res. 29, 99 (2010).
Usuda, J. et al. Klotho predicts good clinical outcome in patients with limited-disease small cell lung cancer who received surgery. Lung Cancer. 74, 332–337 (2011).
Usuda, J. et al. Klotho is a novel biomarker for good survival in resected large cell neuroendocrine carcinoma of the lung. Lung Cancer. 72, 355–359 (2011).
Takegahara, K. et al. Antiaging gene Klotho regulates epithelial-mesenchymal transition and increases sensitivity to pemetrexed by inducing lipocalin-2 expression. Oncol. Lett. 21, 418–418 (2021).
Nishimura, T. et al. Current status of clinical proteogenomics in lung cancer. Expert Rev. Proteom. 16, 761–772 (2019).
Demichev, V., Messner, C. B., Vernardis, S. I., Lilley, K. S. & Ralser, M. DIA-NN: Neural networks and interference correction enable deep proteome coverage in high throughput. Nat. Methods. 17, 41–44 (2020).
Langfelder, P. & Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 9, 559 (2008).
Nishimura, T. et al. Protein co-expression network-based profiles revealed from laser-microdissected cancerous cells of lung squamous-cell carcinomas. Sci. Rep. 11, 20209 (2021).
Krämer, A., Green, J., Pollard, J. Jr. & Tugendreich, S. Causal analysis approaches in ingenuity pathway analysis. Bioinformatics. 30, 523–530 (2014).
Franz, M. et al. GeneMANIA update 2018. Nucleic Acids Res. 46, W60–W64 (2018).
Szklarczyk, D. et al. The STRING database in 2021: Customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 49, D605–D612 (2021).
Chin, C. H. et al. cytoHubba: Identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol. 8(Suppl 4), S11 (2014).
Junjappa, R. P., Patil, P., Bhattarai, K. R., Kim, H. R. & Chae, H. J. IRE1α implications in endoplasmic reticulum stress-mediated development and pathogenesis of autoimmune diseases. Front. Immunol. 9, 1289 (2018).
Zhou, W. H. et al. The overexpression of fibronectin 1 promotes cancer progression and associated with M2 macrophages polarization in head and neck squamous cell carcinoma patients. Int. J. Gen. Med. 15, 5027–5042 (2022).
Marie, P. J. & Haÿ, E. Cadherins and Wnt signalling: A functional link controlling bone formation. Bonekey Rep. 2, 330 (2013).
Osuka, S. et al. N-cadherin upregulation mediates adaptive radioresistance in glioblastoma. J. Clin. Invest. 131, e136098 (2021).
Liu, L. X. et al. Targeting cadherin-17 inactivates Wnt signaling and inhibits tumor growth in liver carcinoma. Hepatology 50, 1453–1463 (2009).
Liu, J. et al. Wnt/β-catenin signalling: function, biological mechanisms, and therapeutic opportunities. Signal Transduct. Target Ther. 7, 3 (2022).
Lánczky, A. & Győrffy, B. Web-based survival analysis tool tailored for medical research (KMplot): development and implementation. J. Med. Internet Res. 23, e27633 (2021).
Zhu, P. et al. Protocadherin 9 inhibits epithelial-mesenchymal transition and cell migration through activating GSK-3β in hepatocellular carcinoma. Biochem. Biophys. Res. Commun. 452, 567–574 (2014).
(No author listed) The tumor suppressor FAT1 modulates WNT activity in multiple cancers. Cancer Discov. 3, OF19 (2013).
Morris, L. G. et al. Recurrent somatic mutation of FAT1 in multiple human cancers leads to aberrant Wnt activation. Nat. Genet. 45, 253–261 (2013).
Yan, G., Chen, V., Lu, X. & Lu, S. A signal-based method for finding driver modules of breast cancer metastasis to the lung. Sci. Rep. 7, 10023 (2017).
Woods, N. T., Yamaguchi, H., Lee, F. Y., Bhalla, K. N. & Wang, H. G. Anoikis, initiated by Mcl-1 degradation and Bim induction, is deregulated during oncogenesis. Cancer Res. 67, 10744–10752 (2007).
Lelievre, E. C. et al. N-cadherin mediates neuronal cell survival through bim down-regulation. PLoS ONE 7, e33206 (2012).
Wang, Y. & Sun, Z. Current understanding of klotho. Ageing Res. Rev. 8, 43–51 (2009).
Zhou, L., Li, Y., Zhou, D., Tan, R. J. & Liu, Y. Loss of Klotho contributes to kidney injury by derepression of Wnt/β-catenin signaling. J. Am. Soc. Nephrol. 24, 771–785 (2013).
Satoh, M. et al. N. Klotho protects against mouse renal fibrosis by inhibiting Wnt signaling. Am. J. Physiol. Ren. Physiol. 303, 1641–1651 (2012).
Muñoz-Castañeda, J. R. et al. Klotho/FGF23 and Wnt signaling as important players in the comorbidities associated with chronic kidney disease. Toxins Basel 12, 185 (2020).
Lu, M. et al. E-cadherin couples death receptors to the cytoskeleton to regulate apoptosis. Mol. Cell. 54, 987–998 (2014).
Chai, G. et al. A human pleiotropic multiorgan condition caused by deficient Wnt secretion. N. Engl. J. Med. 385, 1292–1301 (2021).
Nakashima, Y. & Omasa, T. What kind of signaling maintains pluripotency and viability in human-induced pluripotent stem cells cultured on laminin-511 with serum-free medium?. Biores. Open Access. 5, 84–93 (2016).
Rachagani, S. et al. MUC4 potentiates invasion and metastasis of pancreatic cancer cells through stabilization of fibroblast growth factor receptor 1. Carcinogenesis 33, 1953–1964 (2012).
Goetz, R. et al. Klotho coreceptors inhibit signaling by paracrine fibroblast growth factor 8 subfamily ligands. Mol. Cell. Biol. 32, 1944–1954 (2012).
Lu, Y. et al. Hypoxia induces resistance to EGFR inhibitors in lung cancer cells via upregulation of FGFR1 and the MAPK pathway. Cancer Res. 80, 4655–4667 (2020).
Knights, V. & Cook, S. J. De-regulated FGF receptors as therapeutic targets in cancer. Pharmacol. Ther. 125, 105–117 (2010).
Buchtova, M. et al. Fibroblast growth factor and canonical WNT/β-catenin signaling cooperate in suppression of chondrocyte differentiation in experimental models of FGFR signaling in cartilage. Biochim. Biophys. Acta. 1852, 839–850 (2015).
Chen, B., Ma, X., Liu, S., Zhao, W. & Wu, J. Inhibition of lung cancer cells growth, motility and induction of apoptosis by Klotho, a novel secreted Wnt antagonist, in a dose-dependent manner. Cancer Biol. Ther. 13, 1221–1228 (2012).
Tang, X. et al. Klotho: a tumor suppressor and modulator of the Wnt/β-catenin pathway in human hepatocellular carcinoma. Lab. Invest. 96, 197–205 (2016).
Ley, R., Balmanno, K., Hadfield, K., Weston, C. & Cook, S. J. Activation of the ERK1/2 signaling pathway promotes phosphorylation and proteasome-dependent degradation of the BH3-only protein. Bim. J. Biol. Chem. 278, 18811–18816 (2003).
Datta, S. R. et al. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell 91, 231–241 (1997).
Balmanno, K. & Cook, S. J. Tumour cell survival signalling by the ERK1/2 pathway. Cell Death Differ. 16, 368–377 (2009).
Gillings, A. S., Balmanno, K., Wiggins, C. M., Johnson, M. & Cook, S. J. Apoptosis and autophagy: BIM as a mediator of tumour cell death in response to oncogene-targeted therapeutics. FEBS J. 276, 6050–6062 (2009).
Zougman, A. & Banks, R. E. C-STrap sample preparation method-in-situ cysteinyl peptide capture for bottom-up proteomics analysis in the STrap format. PLoS One 10, e0138775 (2015).
Elinger, D., Gabashvili, A. & Levin, Y. Suspension trapping (S-trap) is compatible with typical protein extraction buffers and detergents for bottom-up proteomics. J. Proteome Res. 18, 1441–1445 (2019).
Bang, G. et al. Comparison of protein characterization using in solution and S-trap digestion methods for proteomics. Biochem. Biophys. Res. Commun. 589, 197–203 (2022).
Wu, C. et al. Coupling suspension trapping-based sample preparation and data-independent acquisition mass spectrometry for sensitive exosomal proteomic analysis. Anal. Bioanal. Chem. 414, 2585–2595 (2022).
Costanzo, M. et al. Dataset of a comparative proteomics experiment in a methyl malonyl-CoA mutase knockout HEK 293 cell model. Data Brief. 33, 106453 (2020).
Jiang, N. et al. Data-independent acquisition (DIA)-based quantification workflow for proteome analysis of 5000 cells. J. Pharm. Biomed. Anal. 216, 114795 (2022).
Demichev, V. et al. Dia-PASEF data analysis using FragPipe and DIA-NN for deep proteomics of low sample amounts. Nat. Commun. 13, 3944 (2022).
Khan, A. & Mathelier, A. Intervene: A tool for intersection and visualization of multiple gene or genomic region sets. BMC Bioinform. 18, 287 (2017).
Author information
Authors and Affiliations
Contributions
J.U., M.M., and T.N. conceived this study. J.U., Y.B., and T.N. initiated and managed the collaboration. M.M., N.O., and J.U. prepared A549 and A549/KL cell strains, performed the invasion assay, and examined metastatic potential in the tumor-bearing mice model. T.F. performed sample preparation and MS-based proteomic analysis. T.N. designed and performed bioinformatics analysis for protein expression data. N.O. performed western blotting. M.M. and T.N. wrote the main manuscript text and prepared Tables 1 and 2, Figs. 1–6, and Supplementary Information File 1. M.M., T.N., and J.U. wrote the first draft of the manuscript. All authors reviewed the manuscript and agreed with the submission.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Matsumoto, M., Ogawa, N., Fukuda, T. et al. Protein interaction networks characterizing the A549 cells Klotho transfected are associated with activated pro-apoptotic Bim and suppressed Wnt/β-catenin signaling pathway. Sci Rep 14, 2130 (2024). https://doi.org/10.1038/s41598-024-52616-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-52616-0
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
