
Abstract
In multiple neurodegenerative diseases, including Alzheimer’s disease (AD), a prominent pathological feature is the aberrant aggregation and inclusion formation of the microtubule-associated protein tau. Because of the pathological association, these disorders are often referred to as tauopathies. Mutations in the MAPT gene that encodes tau can cause frontotemporal dementia with parkinsonism linked to chromosome 17 (FTDP-17), providing the clearest evidence that tauopathy plays a causal role in neurodegeneration. However, large gaps in our knowledge remain regarding how various FTDP-17-linked tau mutations promote tau aggregation and neurodegeneration, and, more generally, how the tauopathy is linked to neurodegeneration. Herein, we review what is known about how FTDP-17-linked pathogenic MAPT mutations cause disease, with a major focus on the prion-like properties of wild-type and mutant tau proteins. The hypothesized mechanisms by which mutations in the MAPT gene promote tauopathy are quite varied and may not provide definitive insights into how tauopathy arises in the absence of mutation. Further, differences in the ability of tau and mutant tau proteins to support prion-like propagation in various model systems raise questions about the generalizability of this mechanism in various tauopathies. Notably, understanding the mechanisms of tauopathy induction and spread and tau-induced neurodegeneration has important implications for tau-targeting therapeutics.
Similar content being viewed by others


Introduction
The microtubule (MT)-associated protein tau (MAPT) is an intrinsically disordered protein expressed at its highest levels in neurons throughout the central nervous system. Higher molecular mass isoforms generated through alternative splicing, often termed “big tau,” are expressed primarily in the peripheral nervous system, but are sometimes also observed in the spinal cord and skeletal muscle, when exon 4a and exon 6 are translated, respectively [1,2,3]. One of tau’s primary functions is to bind to and promote the assembly and stability of MTs; this binding activity can be negatively regulated by phosphorylation at select sites [1, 4].
Tauopathies refer to a wide range of phenotypically diverse diseases characterized by the aberrant aggregation of tau in neurons and/or glia, including Alzheimer’s disease (AD), progressive supranuclear palsy (PSP), corticobasal degeneration (CBD), Pick’s disease (PiD), chronic traumatic encephalopathy (CTE), and frontotemporal dementia with parkinsonism linked to chromosome 17 (FTDP-17) [5]. First discovered as a MT-associated protein in 1975 [6], tau was later found to be the principal component of neurofibrillary tangles (NFTs), which are hyperphosphorylated proteinaceous inclusions found in AD and other tauopathies [7].
In 1998, autosomal dominant mutations in the MAPT gene that encodes tau were found to cause some forms of FTDP-17 [8,9,10], now referred to as FTDP-17t, proving that tau dysfunction is sufficient for widespread central nervous system neurodegeneration. Disease pathology for individuals with FTDP-17t is characterized by the presence of filamentous tau inclusions throughout the frontal and temporal lobes in neurons and sometimes in glia, accompanied by atrophy in these regions, as well as ventricular dilation [11]. These MAPT mutations can cause variable cognitive, behavioral, and motor deficits, with an average age of onset of 49 years and a duration of disease of 8.5 years [12].
As of 2018, over 50 different pathogenic MAPT missense, silent, and intronic mutations have been reported (Figs. 1 and 2, Table 1) [11]. Because many of these mutations present neuropathologically in a manner consistent with different sporadic tauopathies such as PSP, CBD, and PiD [13,14,15], there have recently been calls to characterize tauopathy caused by certain mutations as familial versions of these specific diseases [16]. Additionally, some of these mutations have been found to be risk modifiers in certain tauopathies, for instance, A152T in AD [17]. In addition to the phenotypic and neuropathological variability between these mutations, there are also a number of mechanisms by which these mutations are thought to cause disease. Loss of function, including MT binding and assembly, changes in alternative splicing, shifts in protein-aggregation kinetics, and, more recently, prion-like “seeding,” have all been implicated. Thus, this review focuses on the potential biochemical and cellular mechanisms in which different tau mutants might cause disease—with an emphasis on their ability to aggregate with seeding—and the implications this might have on the study of sporadic tauopathy.

The longest tau isoform found in human brain, with its corresponding mRNA and known pathogenic missense and deletion mutations. The MAPT mRNA resulting in 2N4R tau is shown with an embedded number corresponding to the originating exon. Exon 1 contains both untranslated 5’ region and the start of the protein. Exons 2 and 3 are present in this isoform as N1 and N2 inserts; however, in the 1N and 0N isoforms of tau, exon 2 or neither exon 2 or 3 is translated, respectively. MT-binding repeat 2, or R2, is present in this isoform; however, in 3R tau, exon 10 is alternatively spliced, and this region is not present in the protein. The different colors serve to highlight the regions of the protein that are alternatively spliced as well as the MT-binding domain. The N- terminal, proline-rich, MT-binding, and C-terminal regions are indicated above. Below the protein, known pathogenic missense mutations are indicated. Many of these mutations reside in the MT-binding region, and as such the specific amino-acid sequence of this area is shown. The PHF6* and PHF6 motifs, which are important for tau aggregation, are also indicated. MT, microtubule

Schematic of the RNA stem loop present at the end of exon 10 and the beginning of intron 10. Known pathogenic exonic (both missense and silent) and intronic mutations with their corresponding amino-acid changes or deletions, when applicable, are shown. The boundary between exon 10 and intron 10 is indicated by the partition at the top left and also by the use of lower case letters for intron 10. Mutations in this region that have been shown to increase exon 10 inclusion are indicated in red, while those that have been shown to decrease exon 10 inclusion are indicated in green. Mutations that have not been shown to affect splicing are represented in black
Tau expression and splicing
The MAPT gene, located on chromosome 17, comprises 16 exons, numbered 0–14 [5]. Exon 1 contains both 5’ untranslated region as well as the start codon of the protein, while exon 14 contains untranslated 3’ region. Splicing variants that include exon 4a are primarily present in the peripheral nervous system, while variants that include exon 6 can be found specifically in the spinal cord and skeletal muscle, resulting in a higher molecular mass protein referred to as “big tau” [1,2,3, 5]. In the human brain, six distinct isoforms of tau exist based on the alternative splicing of exons 2, 3, and 10 [18]. Alternative splicing of exons 2 and 3 yields isoforms with 0, 1, or 2 N-terminal repeats (0N, 1N, 2N), while alternative splicing of exon 10 results in tau with three or four MT-binding repeats in the MT-binding domain (3R or 4R) (Fig. 1). While 0N3R tau is the predominant isoform in fetal brain [19], the overall ratio of 3R and 4R tau isoforms is roughly equal in the adult brain [1, 18, 20], although this ratio can differ in given regions [21]. 0N and 1N tau isoforms comprise 37 and 54% of the total human brain tau, respectively, while 2N tau makes up only 9% of the total tau isoforms [22]. 4R tau isoforms show both increased affinity for MTs as well as greater levels of MT assembly in vitro compared to 3R tau isoforms [23, 24]. Although the role of N-terminal inserts is less clear, they have been implicated in regulating MT stabilization and plasma membrane interactions [25, 26]. Additionally, co-immunoprecipitation studies in mice have shown that 0, 1, and 2N isoforms interact with different proteins preferentially [27] and that there are regional differences in the expression of these isoforms in the brain [28].
Alternative RNA splicing of tau is regulated by several cis elements and trans-acting factors [29]. Exon 10, in particular, is flanked by an abnormally large intron 9 and has a weak 5’ and 3’ splice site, which can be acted upon by these cis elements and trans-acting factors [30]. Additionally, the 3’-end of exon 10 and the 5’-end of intron 10 form a highly self-complementary stem loop (Fig. 2), which inhibits the binding of the U1 small nuclear RNA (snRNA) molecule, part of the small nuclear ribonucleoprotein particle (snRNP) that functions to bind to the pre-mRNA and catalyzes the removal of introns [29]. Disruption of this loop leads to increased binding by the snRNP and higher levels of exon 10 inclusion [30]. This destabilization can also be seen in rodents, in which the presence of a guanine instead of an adenine at position IVS10 + 13 leads to the predominance of 4R tau [29]. Thus, mutations within specific cis elements can promote or suppress the inclusion of this exon, while mutations specifically within the stem loop tend to promote exon 10 inclusion (Fig. 2).
Two major haplotypes of tau, H1 and H2, are formed primarily by the 900 kb inversion in the q21 region of chromosome 17 and a 238 bp deletion in intron 9 in H2 [3]. Furthermore, a number of single-nucleotide polymorphisms in the H1 haplotype produce several sub-variants, some of which are associated with an increased risk of certain tauopathies, such as CBD and PSP [31,32,33]. Mechanistically, it is thought that distinct haplotypes, particularly H1c, can promote tauopathy through the increased expression of 4R tau, as is seen in some FTDP-17t mutant tauopathies [34].
Tau structure and aggregation
Four general domains of tau include the N-terminal acidic projection domain, the proline-rich domain, the MT-binding domain, and the C-terminal tail (Fig. 1). Although tau is intrinsically disordered and natively unfolded [35, 36], it can adopt a “paperclip” like structure, in which the MT-binding domain and the N terminus approach and interact with the C terminus [37]. Preclusion of this structure, through the interaction with other molecules, post-translational modifications, truncations, or mutations, could potentiate abnormal aggregation. The MT-binding repeats also comprise the “paired-helical filament core,” or PHF core, which serves as the primary structure of aggregated tau filaments [38]. Within this structure are two hexapeptide motifs, termed PHF6* and PHF6, which are important for aggregation, and the latter is necessary and sufficient for β-sheet aggregation in tau [39,40,41]. Recent cryo-electron microscopy studies of the PHF core from both AD and PiD tau filaments further confirm the key areas of β-sheet forming residues in this core [42, 43]. Overall, the missense MAPT mutations that disrupt the proposed “paperclip” structure of tau, or promote and stabilize the PHF core of tau, are likely to promote aggregation and inclusion formation.
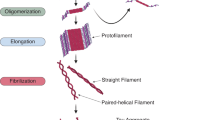
The process by which tau polymerizes to form amyloid in vivo is not completely understood. Nevertheless, “nucleation-elongation” is a potential mechanism that can contribute to this process in which tau initially forms an oligomeric nucleus or “seed” before elongating into tau filaments [44, 45]. As such, the formation of this oligomeric nucleus is the rate-limiting step, after which tau can elongate rapidly by attaching to the growing ends of the fibril [45,46,47]. It has recently been proposed that tau can undergo liquid–liquid separation to form condensed liquid droplets within cellular physiological conditions, which could serve as a precursor for this initial tau β-sheet formation and aggregation [48]. Experimentally, this rate-limiting step can be potentially overcome by introducing the pre-formed “nuclei” of tau that can act as a template for soluble tau to quickly bind to and polymerize onto, in a process known as seeding [49]. This mechanism is akin to the misfolding of prion protein, in which exogenous or intrinsic pathogenic prion conformers act as templates that induce conformational changes in the native protein, inducing misfolding, further aggregation, and neurodegeneration [50]. In a similar manner, it is thought that certain neurodegenerative proteinopathies can spread to anatomically connected regions through template-assisted conformational changes, in which soluble protein is induced to become pathological. This concept is further supported based on AD autopsy series studies, as Braak stages I–VI [51], in which tau pathology in AD can be defined in a regionally specific and somewhat predictable manner. Evidence that tau aggregation can be induced by exogenous tau aggregates and subsequently spread in this manner—through some combinatorial processes of synaptic or exosomal secretion followed by endocytotic or exosomal uptake, for example [49, 52]—has been shown in vivo in various types of cell culture systems and in animal studies (Tables 2–4). Of note, many of these studies heavily utilize specific mutants that may serve to enhance or act as a primer for this process (Tables 2–4).
Tau function and post-translational modifications
Tau resides mostly in the axons of developed neurons, where it has a higher affinity for MTs than in the dendrites [53, 54]. Additionally, different isoforms of tau can have distinct localization patterns, and missorting of tau into dendrites is an early sign of neurodegeneration in AD [53, 55, 56]. Thus, altered splicing patterns may contribute to tau mislocalization and altered MT stability. Normal tau has roles in regulating axonal transport [57] and promoting neurite outgrowth [58]. Knockout mice have further demonstrated important roles for tau in neurogenesis and neuroplasticity, with significant impairments to both in the absence of tau [59, 60]. Tau also binds to and interacts with a number of other molecules. In particular, the N-terminal domain, which has a negative charge and projects away from the MTs when tau is bound [61], can act as a link to membrane components, particularly annexin 2 [62]. This region also binds to the p150 subunit of the dynactin complex, which regulates the MT motor protein dynein [63].
Given that tau functions to enhance MT assembly and regulate its stability, which play important roles in neurite outgrowth, cell stabilization, and intracellular transport, normal tau activity contributes to maintaining axonal health. Tau binds to the interface between α- and β-tubulin heterodimers specifically with residues interspersed throughout and around the MT-binding repeats [64]. Thus, tau mutations within these repeats have the ability to disrupt this interaction, resulting in the destabilization of MTs as well as a potential increase in unbound, free-floating tau, which may also promote tau aggregation (Table 1).
Normally, the process by which tau interacts with MTs is negatively regulated by phosphorylation [4]. There are over 80 potential phosphorylation sites (i.e., serine, threonine, and tyrosine residues) on the longest isoform of tau, a number of which are abnormally hyperphosphorylated in AD and other tauopathies [53]. This hyperphosphorylation may induce the missorting of tau [65], as well as potentially promote aggregation, as shown in vitro [66]. Individual missense mutations in tau can not only alter potential phosphorylation sites, but also have been shown to promote phosphorylation compared to WT tau in vitro [67]. Lysine acetylation has also been shown to be of importance with regard to tau pathology. Depending on the residue, acetylation can inhibit tau’s degradation and correlate with tauopathy, or promote its degradation and suppress aggregation [68, 69]. Other post-translational modifications include glycosylation, isomerization, methylation, ubiquitination, and truncation [70]. N-glycosylation, isomerization, and truncation are implicated in the promotion and stabilization of PHFs [71,72,73], while methylation has been shown in vitro to suppress aggregation [74].
Altered Tau mRNA splicing in disease
Normally, the ratio of 3R to 4R tau in adult human brain is approximately equal [1, 18, 20]. In AD, this ratio remains normal [75]; however, many tauopathies exhibit altered ratios of tau isoforms, especially within the pathologic inclusions. For instance, PSP and CBD are considered 4R tauopathies, while PiD is considered a 3R tauopathy [1]. Interestingly, specific MAPT mutations can cause either 4R or 3R predominant tauopathies as well as tauopathies with equal isoform ratios [76]. Intronic pathogenic mutations largely reside in intron 10 and serve to disrupt the mRNA stem loop, usually causing an increase in 4R tau (Fig. 2). For instance, the most common intronic mutation, IVS10+16 (C to U), serves to increase 4R tau expression by disrupting this stem loop [11], as do a number of other intronic mutations in this area (Fig. 2). Additionally, IVS10+11 (U to C), IVS10+12 (C to U), IVS10+13 (A to G), IVS10+14 (C to U), and IVS10+16 (C to U) reside on an intronic splicing silencer and all increase exon 10 inclusion, while IVS10+19 (C to G) resides on an adjacent intronic splicing modulator and increases exon 10 splicing [77]. Interestingly, the IVS10+13 (A to G) mutation is naturally occurring in rodents, resulting in a preponderance of 4R tau in these animals [29]. Deletion studies have shown that these two regions have opposing effects on splicing [29, 30]. Two missense mutations that affect splicing in opposing ways are N279K and Δ280K, which are both found around a polypurine enhancer, which interacts with a number of different regulatory splicing sequences [77]. While the Δ280K mutation weakens this enhancer, the N279K mutation strengthens it, producing dramatically opposing effects on exon 10 inclusion. The majority of missense mutations that are found in exon 10 are shown to increase the levels of 4R tau expression as revealed by mRNA analysis and/or the protein biochemical profile of patients’ tau isoforms (Table 1). These mutations likely cause disease either through the direct disruption of the mRNA stem loop (Fig. 2) or through the disruption of splicing enhancers and silencers [18, 77]. Furthermore, of the silent mutations found in tau, only those residing in exon 10 (L284L, N296N, or S305S) or exon 11 (L315L) have shown to be pathogenic and could also affect splicing by the disruption of the stem loop (S305S) or by disrupting or enhancing the splicing cis elements (Fig. 2) [77].
Alterations in the normal ratio of tau isoforms can lead to a number of adverse results, including impaired axonal transport [78]. Additionally, for all of the mutations that affect splicing, insoluble tau levels from affected patients’ brains are predominantly but not exclusively comprised the isoform that is over represented (Table 1) [79, 80]. Furthermore, because it is proposed that different tau isoforms harbor distinct MT-binding affinities and potentially bind unique sites, an overproduction of one isoform over another might lead to an overabundance of free-floating, unbound tau, which is primed for aggregation [23, 24, 81].
Altered MT assembly and binding due to tau mutations
Tau missense mutations can affect MT assembly and binding, thus reducing MT stability, as well as potentially leading to increased levels of unbound, free-floating tau in the cytosol of neurons. In fact, one of most common features among missense mutations is their diminished ability, at least in vitro, to promote MT assembly from tubulin compared to WT tau (Table 1). Conversely, a few mutations, such as Q336H and Q336R, have a greater ability to promote MT assembly (Table 1) [82, 83]. Additionally, some missense mutations, such as S305N, that predominantly affect splicing demonstrate little effect on MT polymerization (Table 1) [84]. The impact of tau missense mutations on its interactions with MTs has also been studied in vitro by comparing the binding affinity for taxol-stabilized MTs (Table 1). Although this interaction has been not been as extensively studied as MT assembly, typically, these data demonstrate a reduced MT-binding affinity that is consistent with a reduced ability to induce MT polymerization (Table 1).
Impacts of tau mutations on aggregation and seeding
Mutations that alter splicing dynamics and that induce loss of function with regard to MT interaction both have the potential to increase the amount of free, unbound forms of tau in the cytosol that can ultimately potentiate aggregation and inclusion formation. In vitro, recombinant tau needs a polyanionic inducer, such as heparin or arachidonic acid, in order to form tau filaments [85]. In the cytosol, similar cofactors could promote this aggregation, including RNA [86, 87]. In addition to specific cofactors, mutations in tau may help to induce aggregation, making tau more susceptible to template-assisted growth. In vitro, a number of tau mutants have been shown to increase the rate at which tau fibrillizes (Table 1).
Some tau mutations can also have marked effects on seed-induced tau aggregation in vivo. Growing evidence in cell culture supports prion-like seeding as a possible mechanism that contributes to pathogenesis (Table 2). Original studies conducted have shown that the addition of exogenous pre-formed tau fibrils can induce tau aggregation in cells expressing WT human tau protein [88, 89]. Other studies have focused on the seeding and aggregation of repeat domain (RD) tau, or tau only containing the MT-binding repeats, as these constructs contain the region responsible for tau fibrillization and are more prone to aggregation when seeded or even simply expressed in culture [44, 90,91,92]. The majority of cell culture studies, however, have utilized tau mutants, namely the P301L or P301S mutants, which robustly aggregate with seeding compared to WT tau [91,92,93,94,95,96]. Even when comparing WT RD tau with RD tau containing a P301L or P301S mutation, these differences in aggregation remain [90, 92].
Additionally, the types of tau seeds that were used to treat these tau-expressing cells were largely divided into brain lysate from transgenic mice expressing human tau with a P301L or P301S mutation, human brain lysate from patients with tauopathy, or recombinant tau protein fibrillized in vitro using a polyanionic inducer (Table 2). Comparative studies have shown that tau from brain lysates are more potent at inducing aggregation than recombinant fibrils; however, recombinant tau that was “seeded” in cell culture by detergent-insoluble tau from brain lysate can acquire this potency [94].
Evidence for seeding in vivo has been shown in a number of mouse models (Tables 3 and 4), beginning with Clavaguera et al., where pathology was found at the site of injection with insoluble P301S brain extract in human WT-overexpressing mice 6 months post-injection, with limited spread to nearby regions thereafter [97]. Subsequent studies have mostly utilized mice expressing human mutant P301L or P301S tau, which have shown significant seeding and synaptic spread of pathology with a variety of “seeds” utilized (Tables 3 and 4). These mice were treated, largely, through stereotactic hippocampal injection at 2–3 months, with brain homogenates from different tauopathies [98], lysate from aged P301L or P301S tau-expressing mice [99, 100], lysate from cells with stably expressing tau aggregates [92, 101], or recombinant fibrils, usually the K18 tau fragment (residues 244–372) [102,103,104,105,106].
Brain lysates from human tauopathy cases or from aged mutant tau transgenic mice containing tau aggregates appear to generate the most potent seeds [94]. Further, using sarkosyl-insoluble lysate or immunoprecipitating tau from these brains rather than total lysate appears to enhance their potency [94], while immunodepleting tau greatly diminishes this potency [100]. This, along with evidence that “strains” of tau aggregates can survive multiple passages through both cell culture and mice [101], points to the importance of conformational templating in this process. Isoform differences could also contribute to differential templating; because the P301 residue only exists in 4R tau and because murine tau is almost exclusively 4R [19, 53], most studies have only compared seeding of 4R-expressed tau with 4R aggregates. This differs from the physiological conditions of AD and PiD, as well as from many instances of FTDP-17t. In fact, when PiD brain lysates, which predominately contain 3R tau, were used to seed 4R tau in vivo, there was significantly less pathology [107]. This apparent seeding barrier has also been seen in vitro and in cell culture [89, 108,109,110,111], and corroborates the different proposed structures of tau filaments from AD and PiD elucidated by cryo-electron microscopy [42, 43].
Differences in the entity of seeds comprising either WT or mutant tau appear to matter less than the methods used to generate the seeds (Tables 2–4). For the monomer to be seeded, however, the data in both cell culture and in vivo show the importance of whether WT tau or mutant tau is utilized. Specifically, studies have shown consistent differences in seeding and aggregation propensities between P301L, P301S, and P301T mutant and WT tau in vitro, in cell culture, and in mouse models [90, 92, 94,95,96, 109, 112, 113].
A recent study from our laboratory investigated 19 different pathogenic tau missense mutations in the context of an established cellular seeding assay [109]. It was found that the majority of mutants, including WT tau, failed to robustly aggregate with seeding, unlike tau with mutations at the P301 site. This pattern of aggregation with seeding was similar among the known FTDP-17t mutants P301L, P301S, and P301T, and even a deletion at the P301 site; additionally, there was no difference in seeding between the 0N4R or 2N4R isoforms [109]. Because prolines can serve as inhibitors of β-sheet formation [114], the impact of other proline residues throughout the MT-binding region was investigated; it was found again that the inhibitory effect of P301 on aggregation and seeding was unique, but that re-introducing a proline at residue 302 was sufficient to inhibit seeded aggregation [109], demonstrating the importance of the local molecular environment.
Another unique mutant that affected tau aggregation was S320F, which was also able to aggregate with seeding in this assay and even showed some ability to aggregate without seeding [109]. Other studies have shown that this mutant is prone to aggregate in vitro and in vivo compared to WT and other tau mutants [112, 115]. More specifically, this mutation has conferred greater rates of tau nucleation, leading to the production of short fibrils [112], which could explain this mutant's ability to aggregate without the addition of pre-formed fibrils, since the lag time to create nucleated “seeds” is shorter than that for other tau mutants. Mechanistically, this mutation could promote aggregation in a number of different ways. First, while cryo-electron microscopy findings of tau from AD PHFs have shown that, within the amyloidogenic fold, the S320 residue could reside within a hydrophobic pocket [42], the same group found that in PiD tau filaments, cryo-electron microscopy showed S320 to most likely reside within a hydrophobic pocket [43]; thus, the S to F mutation could act by stabilizing this structure. Agreeing with the proposed structure of PiD tau filaments, S320F has been neuropathologically compared to PiD, with an abundance of Pick bodies found in a carrier’s brain [116]. Additionally, despite being intrinsically disordered, tau can adopt a protective paperclip-like global conformation, in which the MT-binding repeat and C-terminal and N-terminal domains approach each other [37]. The substitution of a large, aromatic side chain in the region where the C-terminal and MT-binding domains interact could potentially disrupt this fold, facilitating polymerization. This disruption of the paperclip-like structure of tau has been shown in vitro with pseudo-phosphorylation of tau at the AT8 and PHF1 epitopes, which are markers of tau pathology [117]. Given these two mutants’ unique properties, it was also found that a double-mutant combination of either P301L or P301S with S320F resulted in a rapid and robust aggregation even without seeding [109].
The presence of aggregated tau inclusions remains a constant between MAPT mutant cases, despite the potentially different mechanisms that led to those inclusions. The exact source of neurodegeneration, however, is unclear and could be due to a number of factors. In AD, the rate of cognitive decline correlates positively with the number of NFTs in the brain [118], which could cause neurodegeneration through the disruption of cytoplasmic organelles or by blocking axonal transport [119, 120]. However, evidence in human AD cases has shown that, while correlative, the number of NFTs can far exceed neuronal loss, and in rodent models, many neurons have been shown to die without ever forming NFTs [121,122,123]. Soluble oligomeric species of tau have also been implicated in cellular toxicity when added in culture as well as accelerated pathology when injected into the hippocampus of transgenic mice [124, 125]; however, the mechanisms of potential toxicity remain unclear. Finally, unbound, aggregate-prone, or aggregated tau are unable to perform their normal functions, namely maintaining or assembling MTs; this, in itself, could lead to neurodegeneration through MT disassembly and impaired axonal transport.
Conclusions
Mutations in the MAPT gene can exert several different effects on the functions and properties of tau. These effects can overlap or be completely distinct between mutations, but all result in the formation of aggregated tau inclusions with neuronal loss and atrophy. Mutations that affect tau mRNA splicing can alter the ratio of tau isoforms and lead to the potential dysregulation of MT dynamics as well as an isoform-specific overabundance of soluble, free-floating tau. Mutations that functionally affect tau’s ability to bind to, promote the assembly of or stabilize MTs may also lead to neurodegeneration in a similar manner: through the dysregulation of MT dynamics and/or tau mislocalization, leading to an increase in MT-unattached tau. In both of these instances, this increased amount of unbound tau could increase the usually minute chance of nucleation events occurring, or these proteins could interact with polyanionic molecules in the cytosol, leading to an eventual cascade of elongation and seeding. For other mutations that increase aggregation propensity and/or seeding, perhaps they function under a similar mechanism, but with a higher susceptibility to β-sheet formation and aggregation, speeding up the pathological process. Importantly, impaired MT function and aberrant tau aggregation are not mutually exclusive pathogenic mechanisms. However, it is noteworthy that the observed differences in seeding propensities between different tau mutants do not correlate with an earlier age of onset or shorter duration of disease [12, 116], suggesting that tau loss of function, such as impacts on MT activities or perhaps some still undiscovered functions of tau, could be more important for pathogenesis than a relative increase in aberrant aggregation propensity.
Given the wide-ranging and unique differences between tau mutants, the choice of models used is important. The question remains, however, as to whether utilizing specific tau mutants—namely those at the P301 residue—in the context of aggregation and seeding studies constitutes a model mechanistically similar and applicable to other mutations in FTD and/or sporadic tauopathy: an important fact to consider when utilizing these models to demonstrate therapeutic efficacy.
References
Arendt T, Stieler JT, Holzer M. Tau and tauopathies. Brain Res Bull. 2016;126:238–92.
Couchie D, Mavilia C, Georgieff IS, Liem RK, Shelanski ML, Nunez J. Primary structure of high molecular weight tau present in the peripheral nervous system. Proc Natl Acad Sci USA. 1992;89:4378–81.
Caillet-Boudin M-L, Buée L, Sergeant N, Lefebvre B. Regulation of human MAPT gene expression. Mol Neurodegener. 2015;10:1–14.
Drechsel DN, Hyman AA, Cobb MH, Kirschner MW. Modulation of the dynamic instability of tubulin assembly by the microtubule-associated protein tau. Mol Biol Cell. 1992;3:1141–54.
Guo T, Noble W, Hanger DP. Roles of tau protein in health and disease. Acta Neuropathol. 2017;133:665–704.
Weingarten MD, Lockwood AH, Hwo SY, Kirschner MW. A protein factor essential for microtubule assembly. Proc Natl Acad Sci USA. 1975;72:1858–62.
Grundke-Iqbal I, Iqbal K, Tung YC, Quinlan M, Wisniewski HM, Binder LI. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci USA. 1986;83:4913–7.
Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, et al. Association of missense and 5’-splice-site mutations in tau with the inherited dementia FTDP-17. Nature. 1998;393:702–5.
Poorkaj P, Bird TD, Wijsman E, Nemens E, Garruto RM, Anderson L, et al. Tau is a candidate gene for chromosome 17 frontotemporal dementia. Ann Neurol. 1998;43:815–25.
Spillantini MG, Murrell JR, Goedert M, Farlow MR, Klug A, Ghetti B. Mutation in the tau gene in familial multiple system tauopathy with presenile dementia. Proc Natl Acad Sci USA. 1998;95:7737–41.
Ghetti B, Oblak AL, Boeve BF, Johnson KA, Dickerson BC, Goedert M. Invited review: Frontotemporal dementia caused by microtubule-associated protein tau gene (MAPT) mutations: A chameleon for neuropathology and neuroimaging. Neuropathol Appl Neurobiol. 2015;41:24–46.
Reed LA, Wszolek ZK, Hutton M. Phenotypic correlations in FTDP-17. Neurobiol Aging. 2001;22:89–107.
Poorkaj P, Muma NA, Zhukareva V, Cochran EJ, Shannon KM, Hurtig H, et al. An R5L τ mutation in a subject with a progressive supranuclear palsy phenotype. Ann Neurol. 2002;52:511–6.
Kouri N, Carlomagno Y, Baker M, Liesinger AM, Caselli RJ, Wszolek ZK, et al. Novel mutation in MAPT exon 13 (p.N410H) causes corticobasal degeneration. Acta Neuropathol. 2014;127:271–82.
Rizzini C, Goedert M, Hodges JR, Smith MJ, Jakes R, Hills R, et al. Tau gene mutation K257T causes a tauopathy similar to Pick’s disease. J Neuropathol Exp Neurol. 2000;59:990–1001.
Forrest SL, Kril JJ, Stevens CH, Kwok JB, Hallupp M, Kim WS, et al. Retiring the term FTDP-17 as MAPT mutations are genetic forms of sporadic frontotemporal tauopathies. Brain. 2017;141:521–34.
Coppola G, Chinnathambi S, Lee JJY, Dombroski BA, Baker MC, Soto-ortolaza AI, et al. Evidence for a role of the rare p.A152T variant in mapt in increasing the risk for FTD-spectrum and Alzheimer’s diseases. Hum Mol Genet. 2012;21:3500–12.
Liu F, Gong C-X. Tau exon 10 alternative splicing and tauopathies. Mol Neurodegener. 2008;3:8.
Kosik KS, Orecchio LD, Bakalis S, Neve RL. Developmentally regulated expression of specific tau sequences. Neuron. 1989;2:1389–97.
Goedert M, Spillantini MG, Jakes R, Rutherford D, Crowther RA. Multiple isoforms of human microtubule-associated protein tau: sequences and localization in neurofibrillary tangles of Alzheimer’s disease. Neuron. 1989;3:519–26.
Majounie E, Cross W, Newsway V, Dillman A, Vandrovcova J, Morris CM, et al. Variation in tau isoform expression in different brain regions and disease states. Neurobiol Aging. 2013;34:1922.e7–1922.e12.
Goedert M, Jakes R. Expression of separate isoforms of human tau protein: correlation with the tau pattern in brain and effects on tubulin polymerization. EMBO J. 1990;9:4225–30.
Goode BL, Chau M, Denis PE, Feinstein SC. Structural and functional differences between 3-repeat and 4-repeat tau isoforms: Implications for normal tau function and the onset of neurodegenerative disease. J Biol Chem. 2000;275:38182–9.
Lu M, Kosik KS. Competition for microtubule-binding with dual expression of tau missense and splice isoforms. Mol Biol Cell. 2001;12:171–84.
Niblock M, Gallo J. Tau alternative splicing in familial and sporadic tauopathies. Biochem Soc Trans. 2012;40:677–80.
Derisbourg M, Leghay C, Chiappetta G, Fernandez-Gomez FJ, Laurent C, Demeyer D, et al. Role of the Tau N-terminal region in microtubule stabilization revealed by new endogenous truncated forms. Sci Rep. 2015;5:9659.
Liu C, Song X, Nisbet R, Götz J. Co-immunoprecipitation with Tau isoform-specific antibodies reveals distinct protein interactions and highlights a putative role for 2N Tau in disease. J Biol Chem. 2016;291:8173–88.
Trabzuni D, Wray S, Vandrovcova J, Ramasamy A, Walker R, Smith C, et al. MAPT expression and splicing is differentially regulated by brain region: Relation to genotype and implication for tauopathies. Hum Mol Genet. 2012;21:4094–103.
Qian W, Liu F. Regulation of alternative splicing of tau exon 10. Neurosci Bull. 2014;30:367–77.
D’Souza I, Schellenberg GD. Determinants of 4-repeat tau expression. Coordination between enhancing and inhibitory splicing sequences for exon 10 inclusion. J Biol Chem. 2000;275:17700–9.
Pittman AM, Myers AJ, Duckworth J, Bryden L, Hanson M, Abou-Sleiman P, et al. The structure of the tau haplotype in controls and in progressive supranuclear palsy. Hum Mol Genet. 2004;13:1267–74.
Pittman AM, Fung HC, de Silva R. Untangling the tau gene association with neurodegenerative disorders. Hum Mol Genet. 2006;15:188–95.
Kouri N, Ross OA, Dombroski B, Younkin CS, Serie DJ, Soto-Ortolaza A, et al. Genome-wide association study of corticobasal degeneration identifies risk variants shared with progressive supranuclear palsy. Nat Commun. 2015;6:7247.
Myers AJ, Pittman AM, Zhao AS, Rohrer K, Kaleem M, Marlowe L, et al. The MAPT H1c risk haplotype is associated with increased expression of tau and especially of 4 repeat containing transcripts. Neurobiol Dis. 2007;25:561–70.
Mukrasch MD, Bibow S, Korukottu J, Jeganathan S, Biernat J, Griesinger C, et al. Structural polymorphism of 441-residue Tau at single residue resolution. PLoS Biol. 2009;7:0399–414.
Avila J, Jiménez JS, Sayas CL, Bolós M, Zabala JC, Rivas G, et al. Tau structures. Front Aging Neurosci. 2016;8:1–10.
Jeganathan S, Von Bergen M, Brutlach H, Steinhoff HJ, Mandelkow E. Global hairpin folding of tau in solution. Biochemistry. 2006;45:2283–93.
Wischik CM, Novak M, Thøgersen HC, Edwards PC, Runswick MJ, Jakes R, et al. Isolation of a fragment of tau derived from the core of the paired helical filament of Alzheimer disease. Proc Natl Acad Sci USA. 1988;85:4506–10.
Ganguly P, Do TD, Larini L, Lapointe NE, Sercel AJ, Shade MF, et al. Tau assembly: The dominant role of PHF6 (VQIVYK) in microtubule binding region repeat R3. J Phys Chem B. 2015;119:4582–93.
Smit FX, Luiken JA, Bolhuis PG. Primary fibril nucleation of aggregation prone Tau fragments PHF6 and PHF6. J Phys Chem B. 2017;121:3250–61.
von Bergen M, Friedhoff P, Biernat J, Heberle J, Mandelkow EM, Mandelkow E. Assembly of tau protein into Alzheimer paired helical filaments depends on a local sequence motif ((306)VQIVYK(311)) forming beta structure. Proc Natl Acad Sci USA. 2000;97:5129–34.
Fitzpatrick AWP, Falcon B, He S, Murzin AG, Murshudov G, Garringer HJ, et al. Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature. 2017;547:185–90.
Falcon B, Zhang W, Murzin AG, Murshudov G, Garringer HJ, Vidal R, et al. Structures of filaments from Pick’s disease reveal a novel tau protein fold. Nature. 2018;561:137–40.
Von Bergen M, Barghorn S, Biernat J, Mandelkow EM, Mandelkow E. Tau aggregation is driven by a transition from random coil to beta sheet structure. Biochim Biophys Acta. 2005;1739:158–66.
Friedhoff P, von Bergen M, Mandelkow EM, Davies P, Mandelkow E. A nucleated assembly mechanism of Alzheimer paired helical filaments. Proc Natl Acad Sci USA. 1998;95:15712–7.
Barghorn S, Mandelkow E. Toward a unified scheme for the aggregation of tau into Alzheimer paired helical filaments. Biochemistry. 2002;41:14885–96.
Shammas SL, Garcia GA, Kumar S, Kjaergaard M, Horrocks MH, Shivji N, et al. A mechanistic model of tau amyloid aggregation based on direct observation of oligomers. Nat Commun. 2015;6:7025.
Wegmann S, Eftekharzadeh B, Tepper K, Zoltowska KM, Bennett RE, Dujardin S, et al. Tau protein liquid–liquid phase separation can initiate tau aggregation. EMBO J. 2018;37:e98049.
Goedert M, Masuda-Suzukake M, Falcon B. Like prions: The propagation of aggregated tau and α-synuclein in neurodegeneration. Brain. 2017;140:266–78.
Prusiner S. Novel proteinaceous infectious particles cause scrapie. Science. 1982;216:136–44.
Braak H, Braak E. Staging of alzheimer’s disease-related neurofibrillary changes. Neurobiol Aging. 1995;16:271–8.
Ayers JI, Giasson BI, Borchelt DR. Prion-like spreading in tauopathies. Biol Psychiatry. 2018;83:337–46.
Wang Y, Mandelkow E. Tau in physiology and pathology. Nat Rev Neurosci. 2015;17:22–35.
Hirokawa N, Funakoshi T, Sato-Harada R, Kanai Y. Selective stabilization of tau in axons and microtubule-associated protein 2C in cell bodies and dendrites contributes to polarized localization of cytoskeletal proteins in mature neurons. J Cell Biol. 1996;132:667–79.
Hoover BR, Reed MN, Su J, Penrod RD, Kotilinek LA, Grant MK, et al. Tau mislocalization to dendritic spines mediates synaptic dysfunction independently of neurodegeneration. Neuron. 2010;68:1067–81.
Liu C, Götz J. Profiling murine tau with 0N, 1N and 2N isoform-specific antibodies in brain and peripheral organs reveals distinct subcellular localization, with the 1N isoform being enriched in the nucleus. PLoS One. 2013;8:e84849.
Stamer K, Vogel R, Thies E, Mandelkow E, Mandelkow EM. Tau blocks traffic of organelles, neurofilaments, and APP vesicles in neurons and enhances oxidative stress. J Cell Biol. 2002;156:1051–63.
Caceres A, Kosik KS. Inhibition of neurite polarity by tau antisense oligonucleotides in primary cerebellar neurons. Nature. 1990;343:461–3.
Hong X-P, Peng C-X, Wei W, Tian Q, Liu Y-H, Yao X-Q, et al. Essential role of tau phosphorylation in adult hippocampal neurogenesis. Hippocampus. 2010;20:1339–49.
Kimura T, Whitcomb DJ, Jo J, Regan P, Piers T, Heo S, et al. Microtubule-associated protein tau is essential for long-term depression in the hippocampus. Philos Trans R Soc Lond B Biol Sci. 2014;369:20130144.
Hirokawa N, Shiomura Y, Okabe S. Tau proteins: the molecular structure and mode of binding on microtubules. J Cell Biol. 1988;107:1449–59.
Brandt R, Léger J, Lee G. Interaction of tau with the neural plasma membrane mediated by tau’s amino-terminal projection domain. J Cell Biol. 1995;131:1327–40.
Magnani E, Fan J, Gasparini L, Golding M, Williams M, Schiavo G, et al. Interaction of tau protein with the dynactin complex. EMBO J. 2007;26:4546–54.
Kadavath H, Hofele RV, Biernat J, Kumar S, Tepper K, Urlaub H, et al. Tau stabilizes microtubules by binding at the interface between tubulin heterodimers. Proc Natl Acad Sci USA. 2015;112:7501–6.
Thies E, Mandelkow E-M. Missorting of tau in neurons causes degeneration of synapses that can be rescued by the kinase MARK2/Par-1. J Neurosci. 2007;27:2896–907.
Despres C, Byrne C, Qi H, Cantrelle F-X, Huvent I, Chambraud B, et al. Identification of the Tau phosphorylation pattern that drives its aggregation. Proc Natl Acad Sci USA. 2017;114:9080–5.
Alonso ADC, Mederlyova A, Novak M, Grundke-Iqbal I, Iqbal K. Promotion of hyperphosphorylation by frontotemporal dementia tau mutations. J Biol Chem. 2004;279:34873–81.
Min SW, Chen X, Tracy TE, Li Y, Zhou Y, Wang C, et al. Critical role of acetylation in tau-mediated neurodegeneration and cognitive deficits. Nat Med. 2015;21:1154–62.
Irwin DJ, Cohen TJ, Grossman M, Arnold SE, McCarty-Wood E, Van Deerlin VM, et al. Acetylated tau neuropathology in sporadic and hereditary tauopathies. Am J Pathol. 2013;183:344–51.
Martin L, Latypova X, Terro F. Post-translational modifications of tau protein: Implications for Alzheimer’s disease. Neurochem Int. 2011;58:458–71.
Lee S, Shea TB. Caspase-mediated truncation of Tau potentiates aggregation. Int J Alzheimers Dis. 2012;2012:731063.
Kondo A, Shahpasand K, Mannix R, Qiu J, Moncaster J, Chen CH, et al. Antibody against early driver of neurodegeneration cis P-tau blocks brain injury and tauopathy. Nature. 2015;523:431–6.
Wang JZ, Grundke-Iqbal I, Iqbal K. Glycosylation of microtubule-associated protein tau: An abnormal posttranslational modification in Alzheimer’s disease. Nat Med. 1996;2:871–5.
Funk KE, Thomas SN, Schafer KN, Cooper GL, Clark DJ, Yang AJ, et al. Lysine methylation is an endogenous post-translational modification of tau protein in human brain and a modulator of aggregation propensity. Biochem J. 2015;462:77–88.
Goedert M, Spillantini MG, Jakes R, Crowtherp RA, Vanmechelen E, Probst A, et al. Molecular dissection of the paired helical filament. Neurobiol Aging. 1995;16:325–34.
Dickson DW, Kouri N, Murray ME, Josephs KA. Neuropathology of frontotemporal lobar degeneration-Tau (FTLD-Tau). J Mol Neurosci. 2012;45:384–9.
D’Souza I, Schellenberg GD. Regulation of tau isoform expression and dementia. Biochim Biophys Acta. 2005;1739:104–15.
Stoothoff W, Jones PB, Spires-Jones TL, Joyner D, Chhabra E, Bercury K, et al. Differential effect of three-repeat and four-repeat tau on mitochondrial axonal transport. J Neurochem. 2009;111:417–27.
van Swieten JC, Bronner IF, Azmani A, Severijnen L-A, Kamphorst W, Ravid R, et al. The ΔK280 mutation in MAP tau favors Exon 10 skipping in vivo. J Neuropathol Exp Neurol. 2007;66:17–25.
Ogaki K, Li Y, Takanashi M, Ishikawa KI, Kobayashi T, Nonaka T, et al. Analyses of the MAPT, PGRN, and C9orf72 mutations in Japanese patients with FTLD, PSP, and CBS. Parkinsonism Relat Disord. 2013;19:15–20.
Panda D, Samuel JC, Massie M, Feinstein SC, Wilson L. Differential regulation of microtubule dynamics by three- and four-repeat tau: Implications for the onset of neurodegenerative disease. Proc Natl Acad Sci USA. 2003;100:9548–53.
Pickering-Brown SM, Baker M, Nonaka T, Ikeda K, Sharma S, Mackenzie J, et al. Frontotemporal dementia with Pick-type histology associated with Q336R mutation in the tau gene. Brain. 2004;127:1415–26.
Tacik P, DeTure M, Hinkle KM, Lin WL, Sanchez-Contreras M, Carlomagno Y, et al. A novel Tau Mutation in exon 12, P. Q336H, causes hereditary pick disease. J Neuropathol Exp Neurol. 2015;74:1042–52.
D’Souza I, Poorkaj P, Hong M, Nochlin D, Lee VM, Bird TD, et al. Missense and silent tau gene mutations cause frontotemporal dementia with parkinsonism-chromosome 17 type, by affecting multiple alternative RNA splicing regulatory elements. Proc Natl Acad Sci USA. 1999;96:5598–603.
Goedert M, Jakes R, Spillantini MG, Hasegawa M, Smith MJ, Crowther RA. Assembly of microtubule-associated protein tau into Alzheimer-like filaments induced by sulphated glycosaminoglycans. Nature. 1996;383:550–3.
Dinkel PD, Holden MR, Matin N, Margittai M. RNA binds to tau fibrils and sustains template-assisted growth. Biochemistry. 2015;54:4731–40.
Kampers T, Friedhoff P, Biernat J, Mandelkow EM, Mandelkow E. RNA stimulates aggregation of microtubule-associated protein tau into Alzheimer-like paired helical filaments. FEBS Lett. 1996;399:344–9.
Frost B, Jacks RL, Diamond MI. Propagation of tau misfolding from the outside to the inside of a cell. J Biol Chem. 2009;284:12845–52.
Nonaka T, Watanabe ST, Iwatsubo T, Hasegawa M. Seeded aggregation and toxicity of α-synuclein and tau: Cellular models of neurodegenerative diseases. J Biol Chem. 2010;285:34885–98.
Kfoury N, Holmes BB, Jiang H, Holtzman DM, Diamond MI. Trans-cellular propagation of Tau aggregation by fibrillar species. J Biol Chem. 2012;287:19440–51.
Holmes BB, Furman JL, Mahan TE, Yamasaki TR, Mirbaha H, Eades WC, et al. Proteopathic tau seeding predicts tauopathy in vivo. Proc Natl Acad Sci USA. 2014;111:E4376–4385.
Sanders DW, Kaufman SK, DeVos SL, Sharma AM, Mirbaha H, Li A, et al. Distinct tau prion strains propagate in cells and mice and define different tauopathies. Neuron. 2014;82:1271–88.
Guo JL, Lee VMY. Neurofibrillary tangle-like tau pathology induced by synthetic tau fibrils in primary neurons over-expressing mutant tau. FEBS Lett. 2013;587:717–23.
Falcon B, Cavallini A, Angers R, Glover S, Murray TK, Barnham L, et al. Conformation determines the seeding potencies of native and recombinant Tau aggregates. J Biol Chem. 2015;290:1049–65.
Waxman EA, Giasson BI. Induction of intracellular tau aggregation is promoted by α-synuclein seeds and provides novel insights into the hyperphosphorylation of tau. J Neurosci. 2011;31:7604–18.
Guo JL, Lee VMY. Seeding of normal tau by pathological tau conformers drives pathogenesis of Alzheimer-like tangles. J Biol Chem. 2011;286:15317–31.
Clavaguera F, Bolmont T, Crowther RA, Abramowski D, Frank S, Probst A, et al. Transmission and spreading of tauopathy in transgenic mouse brain. Nat Cell Biol. 2009;11:909–13.
Boluda S, Iba M, Zhang B, Raible KM, Lee VMY, Trojanowski JQ. Differential induction and spread of tau pathology in young PS19 tau transgenic mice following intracerebral injections of pathological tau from Alzheimer’s disease or corticobasal degeneration brains. Acta Neuropathol. 2015;129:221–37.
Ahmed Z, Cooper J, Murray TK, Garn K, McNaughton E, Clarke H, et al. A novel in vivo model of tau propagation with rapid and progressive neurofibrillary tangle pathology: The pattern of spread is determined by connectivity, not proximity. Acta Neuropathol. 2014;127:667–83.
Jackson SJ, Kerridge C, Cooper J, Cavallini A, Falcon B, Cella CV, et al. Short fibrils constitute the major species of seed-competent tau in the brains of mice transgenic for human P301S tau. J Neurosci. 2016;36:762–72.
Kaufman SK, Sanders DW, Thomas TL, Ruchinskas AJ, Vaquer-Alicea J, Sharma AM, et al. Tau prion strains dictate patterns of cell pathology, progression rate, and regional vulnerability in vivo. Neuron. 2016;92:796–812.
Iba M, Guo JL, McBride JD, Zhang B, Trojanowski JQ, Lee VM. Synthetic tau fibrils mediate transmission of neurofibrillary tangles in a transgenic mouse model of Alzheimer’s-like tauopathy. J Neurosci. 2013;33:1024–37.
Peeraer E, Bottelbergs A, Van Kolen K, Stancu IC, Vasconcelos B, Mahieu M, et al. Intracerebral injection of preformed synthetic tau fibrils initiates widespread tauopathy and neuronal loss in the brains of tau transgenic mice. Neurobiol Dis. 2015;73:83–95.
Chakrabarty P, Hudson VJ, Sacino AN, Brooks M, D’Alton S, Lewis J, et al. Inefficient induction and spread of seeded tau pathology in P301L mouse model tauopathy suggest inherent physiological barriers to transmission. Acta Neuropathol. 2015;130:303–5.
Stancu IC, Vasconcelos B, Ris L, Wang P, Villers A, Peeraer E, et al. Templated misfolding of Tau by prion-like seeding along neuronal connections impairs neuronal network function and associated behavioral outcomes in Tau transgenic mice. Acta Neuropathol. 2015;129:875–94.
Vasconcelos B, Stancu IC, Buist A, Bird M, Wang P, Vanoosthuyse A, et al. Heterotypic seeding of Tau fibrillization by pre-aggregated Abeta provides potent seeds for prion-like seeding and propagation of Tau-pathology in vivo. Acta Neuropathol. 2016;131:549–69.
Clavaguera F, Akatsu H, Fraser G, Crowther RA, Frank S, Hench J, et al. Brain homogenates from human tauopathies induce tau inclusions in mouse brain. Proc Natl Acad Sci USA. 2013;110:9535–40.
Adams SJ, de Ture MA, McBride M, Dickson DW, Petrucelli L. Three repeat isoforms of tau inhibit assembly of four repeat tau filaments. PLoS One. 2010;5:e10810.
Strang KH, Croft CL, Sorrentino ZA, Chakrabarty P, Golde TE, Giasson BI. Distinct differences in prion-like seeding and aggregation between tau protein variants provide mechanistic insights into tauopathies. J Biol Chem. 2017;293:2408–21.
Siddiqua A, Luo Y, Meyer V, Swanson MA, Yu X, Wei G, et al. Conformational basis for asymmetric seeding barrier in filaments of three- and four-repeat tau. J Am Chem Soc. 2012;134:10271–8.
Woerman AL, Aoyagi A, Patel S, Kazmi SA, Lobach I, Grinberg LT, et al. Tau prions from Alzheimer’s disease and chronic traumatic encephalopathy patients propagate in cultured cells. Proc Natl Acad Sci USA. 2016;113:E8187–96.
Combs B, Gamblin TC. FTDP-17 tau mutations induce distinct effects on aggregation and microtubule interactions. Biochemistry. 2012;51:8597–607.
Verheyen A, Diels A, Dijkmans J, Oyelami T, Meneghello G, Mertens L, et al. Using human iPSC-derived neurons to model TAU aggregation. PLoS One. 2015;10:e0146127.
Monsellier E, Chiti F. Prevention of amyloid-like aggregation as a driving force of protein evolution. EMBO Rep. 2007;8:737–42.
Bardai FH, Wang L, Mutreja Y, Yenjerla M, Gamblin TC, Feany MB. A conserved cytoskeletal signaling cascade mediates neurotoxicity of FTDP-17 tau mutations in vivo. J Neurosci. 2017;38:1550–1517.
Rosso SM, Van Herpen E, Deelen W, Kamphorst W, Severijnen LA, Willemsen R, et al. A novel tau mutation, S320F, causes a tauopathy with inclusions similar to those in Pick’s disease. Ann Neurol. 2002;51:373–6.
Jeganathan S, Hascher A, Chinnathambi S, Biernat J, Mandelkow EM, Mandelkow E. Proline-directed pseudo-phosphorylation at AT8 and PHF1 epitopes induces a compaction of the paperclip folding of tau and generates a pathological (MC-1) conformation. J Biol Chem. 2008;283:32066–76.
Arriagada PV, Growdon JH, Hedley-Whyte ET, Hyman BT. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer’s disease. Neurology. 1992;42:631–631.
Lin WL, Lewis J, Yen SH, Hutton M, Dickson DW. Ultrastructural neuronal pathology in transgenic mice expressing mutant (P301L) human tau. J Neurocytol. 2003;32:1091–105.
Millecamps S, Julien J-P, Pierre U. Axonal transport deficits and neurodegenerative diseases. Nat Rev Neurosci. 2013;14:161–76.
Gómez-Isla T, Hollister R, West H, Mui S, Growdon JH, Petersen RC, et al. Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer’s disease. Ann Neurol. 1997;41:17–24.
Andorfer C. Cell-cycle reentry and cell death in transgenic mice expressing nonmutant human tau isoforms. J Neurosci. 2005;25:5446–54.
Spires-Jones TL, de Calignon A, Matsui T, Zehr C, Pitstick R, Wu H-Y, et al. In vivo imaging reveals dissociation between caspase activation and acute neuronal death in tangle-bearing neurons. J Neurosci. 2008;28:862–7.
Gerson J, Castillo-Carranza DL, Sengupta U, Bodani R, Prough DS, DeWitt DS, et al. Tau oligomers derived from traumatic brain injury cause cognitive impairment and accelerate onset of pathology in Htau mice. J Neurotrauma. 2016;33:2034–43.
Flach K, Hilbrich I, Schiffmann A, Gärtner U, Krüger M, Leonhardt M, et al. Tau oligomers impair artificial membrane integrity and cellular viability. J Biol Chem. 2012;287:43223–33.
Hayashi S, Toyoshima Y, Hasegawa M, Umeda Y, Wakabayashi K, Tokiguchi S, et al. Late-onset frontotemporal dementia with a novel exon 1 (Arg5His) tau gene mutation. Ann Neurol. 2002;51:525–30.
Chang E, Kim S, Yin H, Nagaraja HN, Kuret J. Pathogenic missense MAPT mutations differentially modulate tau aggregation propensity at nucleation and extension steps. J Neurochem. 2008;107:1113–23.
Iyer A, LaPointe NE, Zielke K, Berdynski M, Guzman E, Barczak A, et al. A novel MAPT mutation, G55R, in a frontotemporal dementia patient leads to altered tau function. PLoS One. 2013;8:e76409.
Silva MC, Cheng C, Mair W, Almeida S, Fong H, Biswas MHU, et al. Human iPSC-derived neuronal model of Tau-A152T frontotemporal dementia reveals tau-mediated mechanisms of neuronal vulnerability. Stem Cell Reports. 2016;7:325–40.
Kara E, Ling H, Pittman AM, Shaw K, de Silva R, Simone R, et al. The MAPT p.A152T variant is a risk factor associated with tauopathies with atypical clinical and neuropathological features. Neurobiol Aging. 2012;33:2231.e7–2231.e14.
Grover A, England E, Baker M, Sahara N, Adamson J, Granger B, et al. A novel tau mutation in exon 9 (I260V) causes a four-repeat tauopathy. Exp Neurol. 2003;184:131–40.
Pickering-Brown S, Baker M, Yen SH, Liu WK, Hasegawa M, Cairns N, et al. Pick’s disease is associated with mutations in the tau gene. Ann Neurol. 2000;48:859–67.
Hogg M, Grujic ZM, Baker M, Demirci S, Guillozet AL, Sweet AP, et al. The L266V tau mutation is associated with frontotemporal dementia and Pick-like 3R and 4R tauopathy. Acta Neuropathol. 2003;106:323–36.
Kobayashi T, Ota S, Tanaka K, Ito Y, Hasegawa M, Umeda Y, et al. A novel L266V mutation of the tau gene causes frontotemporal dementia with a unique tau pathology. Ann Neurol. 2003;53:133–7.
Barghorn S, Zheng-Fischhofer Q, Ackmann M, Biernat J, Von Bergen M, Mandelkow EM, et al. Structure, microtubule interactions, and paired helical filament aggregation by tau mutants of frontotemporal dementias. Biochemistry. 2000;39:11714–21.
Bronner IF, Ter Meulen BC, Azmani A, Severijnen LA, Willemsen R, Kamphorst W, et al. Hereditary Pick’s disease with the G272V tau mutation shows predominant three-repeat tau pathology. Brain. 2005;128:2645–53.
Hasegawa M, Smith MJ, Iijima M, Tabira T, Goedert M. FTDP-17 mutations N279K and S305N in tau produce increased splicing of exon 10. FEBS Lett. 1999;443:93–6.
Wren MC, Zhao J, Liu C-C, Murray ME, Atagi Y, Davis MD, et al. Frontotemporal dementia-associated N279K tau mutant disrupts subcellular vesicle trafficking and induces cellular stress in iPSC-derived neural stem cells. Mol Neurodegener. 2015;10:46.
Hong M, Zhukareva V, Vogelsberg-Ragaglia V, Wszolek Z, Reed L, Miller BI, et al. Mutation-specific functional impairments in distinct tau isoforms of hereditary FTDP-17. Science. 1998;282:1914–7.
Goedert M, Jakes R, Crowther RA. Effects of frontotemporal dementia FTDP-17 mutations on heparin-induced assembly of tau filaments. FEBS Lett. 1999;450:306–11.
Rizzu P, Van Swieten JC, Joosse M, Hasegawa M, Stevens M, Tibben A, et al. High prevalence of mutations in the microtubule-associated protein tau in a population study of frontotemporal dementia in the Netherlands. Am J Hum Genet. 1999;64:414–21.
Von Bergen M, Barghorn S, Li L, Marx A, Biernat J, Mandelkow EM, et al. Mutations of tau protein in frontotemporal dementia promote aggregation of paired helical filaments by enhancing local β-structure. J Biol Chem. 2001;276:48165–74.
Yoshida H, Crowther RA, Goedert M. Functional effects of tau gene mutations deltaN296 and N296H. J Neurochem. 2002;80:548–51.
Grover A, DeTure M, Yen SH, Hutton M. Effects on splicing and protein function of three mutations in codon N296 of tau in vitro. Neurosci Lett. 2002;323:33–6.
Iovino M, Pfisterer U, Holton JL, Lashley T, Swingler RJ, Calo L, et al. The novel MAPT mutation K298E: Mechanisms of mutant tau toxicity, brain pathology and tau expression in induced fibroblast-derived neurons. Acta Neuropathol. 2014;127:283–95.
Nacharaju P, Lewis J, Easson C, Yen S, Hackett J, Hutton M, et al. Accelerated filament formation from tau protein with specific FTDP-17 missense mutations. FEBS Lett. 1999;447:195–9.
Hasegawa M, Smith MJ, Goedert M. Tau proteins with FTDP-17 mutations have a reduced ability to promote microtubule assembly. FEBS Lett. 1998;437:207–10.
Dayanandan R, Van Slegtenhorst M, Mack TGA, Ko L, Yen SH, Leroy K, et al. Mutations in tau reduce its microtubule binding properties in intact cells and affect its phosphorylation. FEBS Lett. 1999;446:228–32.
Elbaum-garfinkle S, Cobb G, Compton JT, Li X-H, Rhoades E. Tau mutants bind tubulin heterodimers with enhanced affinity. Proc Natl Acad Sci USA. 2014;111:6311–6.
Brandt R, Hundelt M, Shahani N. Tau alteration and neuronal degeneration in tauopathies: Mechanisms and models. Biochim Biophys Acta. 2005;1739:331–54.
Bugiani O, Murrell JR, Giaccone G, Hasegawa M, Ghigo G, Tabaton M, et al. Frontotemporal dementia and corticobasal degeneration in a family with a P301S mutation in tau. J Neuropathol Exp Neurol. 1999;58:667–77.
Lladó A, Ezquerra M, Sánchez-Valle R, Rami L, Tolosa E, Molinuevo JL. A novel MAPT mutation (P301T) associated with familial frontotemporal dementia. Eur J Neurol. 2007;14:9–10.
Ros R, Thobois S, Streichenberger N, Kopp N, Sánchez MP, Pérez M, et al. A new mutation of the τ gene, G303V, in early-onset familial progressive supranuclear palsy. Arch Neurol. 2005;62:1444–50.
Kovacs GG, Pittman A, Revesz T, Luk C, Lees A, Kiss E, et al. MAPT S305I mutation: implications for argyrophilic grain disease. Acta Neuropathol. 2008;116:103–18.
Iijima M, Tabira T, Poorkaj P, Schellenberg GD, Trojanowski JQ, Lee VMY, et al. A distinct familial presenile dementia with a novel missense mutation in the tau gene. Neuroreport. 1999;10:497–501.
Van Herpen E, Rosso SM, Serverijnen LA, Yoshida H, Breedveld G, Van De Graaf R, et al. Variable phenotypic expression and extensive tau pathology in two families with the novel tau mutation L315R. Ann Neurol. 2003;54:573–81.
Zarranz JJ, Ferrer I, Lezcano E, Forcadas MI, Eizaguirre B, Atarés B, et al. A novel mutation (K317M) in the MAPT gene causes FTDP and motor neuron disease. Neurology. 2005;64:1578–85.
Tacik P, DeTure M, Lin WL, Sanchez Contreras M, Wojtas A, Hinkle KM, et al. A novel tau mutation, p.K317N, causes globular glial tauopathy. Acta Neuropathol. 2015;130:199–214.
Deramecourt V, Lebert F, Maurage CA, Fernandez-Gomez FJ, Dujardin S, Colin M, et al. Clinical, neuropathological, and biochemical characterization of the novel tau mutation P332S. J Alzheimers Dis. 2012;31:741–9.
Spina S, Murrell JR, Yoshida H, Ghetti B, Bermingham N, Sweeney B, et al. The novel Tau mutation G335S: Clinical, neuropathological and molecular characterization. Acta Neuropathol. 2007;113:461–70.
Neumann M, Diekmann S, Bertsch U, Vanmassenhove B, Bogerts B, Kretzschmar HA. Novel G335V mutation in the tau gene associated with early onset familial frontotemporal dementia. Neurogenetics. 2005;6:91–5.
Deture M, Ko LW, Yen S, Nacharaju P, Easson C, Lewis J, et al. Missense tau mutations identified in FTDP-17 have a small effect on tau-microtubule interactions. Brain Res. 2000;853:5–14.
Spillantini MG, Crowther RA, Goedert M. Comparison of the neurofibrillary pathology in Alzheimer’s disease and familial presenile dementia with tangles. Acta Neuropathol. 1996;92:42–8.
Lippa CF, Zhukareva V, Kawarai T, Uryu K, Shafiq M, Nee LE, et al. Frontotemporal dementia with novel tau pathology and a Glu342Val tau mutation. Ann Neurol. 2000;48:850–8.
Nicholl DJ, Greenstone MA, Clarke CE, Rizzu P, Crooks D, Crowe A, et al. An English kindred with a novel recessive tauopathy and respiratory failure. Ann Neurol. 2003;54:682–6.
Momeni P, Wickremaratchi MM, Bell J, Arnold R, Beer R, Hardy J, et al. Familial early onset frontotemporal dementia caused by a novel S356T MAPT mutation, initially diagnosed as schizophrenia. Clin Neurol Neurosurg. 2010;112:917–20.
Rossi G, Bastone A, Piccoli E, Mazzoleni G, Morbin M, Uggetti A, et al. New mutations in MAPT gene causing frontotemporal lobar degeneration: Biochemical and structural characterization. Neurobiol Aging. 2012;33:834.e1–834.e6.
Popović M, Fabjan A, Mraz J, Magdič J, Glavač D, Zupan A, et al. Tau protein mutation P364S in two sisters: Clinical course and neuropathology with emphasis on new, composite neuronal tau inclusions. Acta Neuropathol. 2014;128:155–7.
Neumann M, Schulz-Schaeffer W, Crowther RA, Smith MJ, Spillantini MG, Goedert M, et al. Pick’s disease associated with the novel Tau gene mutation K369I. Ann Neurol. 2001;50:503–13.
Tacik P, DeTure MA, Carlomagno Y, Lin WL, Murray ME, Baker MC, et al. FTDP-17 with Pick body-like inclusions associated with a novel tau mutation, p.E372G. Brain Pathol. 2017;27:612–26.
Ghetti B, Murrell JR, Zolo P, Spillantini MG, Goedert M. Progress in hereditary tauopathies: A mutation in the Tau gene (G389R) causes a pick disease-like syndrome. Ann N Y Acad Sci. 2000;920:52–62.
Murrell JR, Spillantini MG, Zolo P, Guazzelli M, Smith MJ, Hasegawa M, et al. Tau gene mutation G389R causes a tauopathy with abundant pick body-like inclusions and axonal deposits. J Neuropathol Exp Neurol. 1999;58:1207–26.
Santa-Maria I, Varghese M, Ksiezak-Reding H, Dzhun A, Wang J, Pasinetti GM. Paired helical filaments from Alzheimer disease brain induce intracellular accumulation of tau protein in aggresomes. J Biol Chem. 2012;287:20522–33.
Guo JL, Covell DJ, Daniels JP, Iba M, Stieber A, Zhang B, et al. Distinct α-synuclein strains differentially promote tau inclusions in neurons. Cell. 2013;154:103–17.
Mirbaha H, Holmes BB, Sanders DW, Bieschke J, Diamond MI. Tau trimers are the minimal propagation unit spontaneously internalized to seed intracellular aggregation. J Biol Chem. 2015;290:14893–903.
Audouard E, Houben S, Masaracchia C, Yilmaz Z, Suain V, Authelet M, et al. High molecular-weight paired helical filaments from Alzheimer brain induces seeding of wild-type mouse tau into an argyrophilic 4R tau pathology in vivo. Am J Pathol. 2016;186:2709–22.
Furman JL, Vaquer-Alicea J, White CL, Cairns NJ, Nelson PT, Diamond MI. Widespread tau seeding activity at early Braak stages. Acta Neuropathol. 2017;133:91–100.
Kaufman SK, Thomas TL, Del Tredici K, Braak H, Diamond MI. Characterization of tau prion seeding activity and strains from formaldehyde-fixed tissue. Acta Neuropathol Commun. 2017;5:41.
Devos SL, Corjuc BT, Oakley DH, Nobuhara CK, Bannon RN, Chase A, et al. Synaptic tau seeding precedes tau pathology in human Alzheimer’s disease brain. Front Neurosci. 2018;12:267.
Clavaguera F, Hench J, Lavenir I, Schweighauser G, Frank S, Goedert M, et al. Peripheral administration of tau aggregates triggers intracerebral tauopathy in transgenic mice. Acta Neuropathol. 2014;127:299–301.
Gibbons XGS, Banks RA, Kim B, Xu H, Changolkar L, Leight SN, et al. GFP-mutant human tau transgenic mice develop tauopathy following CNS injections of Alzheimer’s brain-derived pathological tau or synthetic mutant human tau fibrils. J Neurosci. 2017;37:11485–94.
Narasimhan XS, Guo JL, Changolkar L, Stieber A, Mcbride XD, Silva LV, et al. Pathological tau strains from human brains recapitulate the diversity of tauopathies in nontransgenic mouse brain. J Neurosci. 2017;37:11406–23.
Iba M, McBride JD, Guo JL, Zhang B, Trojanowski JQ, Lee VMY. Tau pathology spread in PS19 tau transgenic mice following locus coeruleus (LC) injections of synthetic tau fibrils is determined by the LC’s afferent and efferent connections. Acta Neuropathol. 2015;130:349–62.
Acknowledgements
This work was supported by grants from the National Institutes of Health (R01NS089022) and the Florida Department of Health (7AZ25).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Strang, K.H., Golde, T.E. & Giasson, B.I. MAPT mutations, tauopathy, and mechanisms of neurodegeneration. Lab Invest 99, 912–928 (2019). https://doi.org/10.1038/s41374-019-0197-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41374-019-0197-x
This article is cited by
-
Brain clearance of protein aggregates: a close-up on astrocytes
Molecular Neurodegeneration (2024)
-
Native-state proteomics of Parvalbumin interneurons identifies unique molecular signatures and vulnerabilities to early Alzheimer’s pathology
Nature Communications (2024)
-
Mathematical topology and geometry-based classification of tauopathies
Scientific Reports (2024)
-
Parkinson-ALS with a novel MAPT variant
Neurological Sciences (2024)
-
Clinical and neuroimaging characterization of the first frontotemporal dementia family carrying the MAPT p.K298E mutation
Neurogenetics (2024)
