

Key Points
- Dysphagia is often underestimated in neuromuscular disorders. It can be prominent in dystrophies, inflammatory myopathy, mitochondrial myopathy, myasthenia, motor neuron diseases, and peripheral neuropathy.
- Among muscular dystrophies, oculopharyngeal muscular dystrophy and myotonic dystrophy are most commonly associated with dysphagia.
- Among inflammatory myopathies, the polymyositis associated with connective tissue disease and inclusion body myositis are most likely associated with dysphagia.
- Myasthenia leads to dysphagia with significant oral, masticatory, and pharyngeal components.
- Among motor neuron disorders, amyotrophic lateral sclerosis (ALS) is a leading cause of swallowing impairment, even in the absence of overt bulbar symptoms.
- Swallowing is usually intact in peripheral nerve disorders, except for polyradiculoneuropathies.
Introduction
Patients with progressive difficulties of swallowing present a challenge to the gastroenterologist, otolaryngologist, and neurologist. Besides adding a significant level of disability, certain neuromuscular conditions leading to dysphagia such as amyotrophic lateral sclerosis (ALS) can be fatal, and bulbar presentation may be the predominant feature, requiring a diligent workup and intervention to prevent unwanted complications such as aspiration pneumonia. Feeding problems in neuromuscular disorders are often underestimated. In a survey of 451 patients with 409 responders, the overall prevalence of feeding disability (oral or pharyngeal) was 35%.1 A better understanding of the swallowing problems associated with these disorders may help in guiding treatment, choosing technical aids, modifying the consistency of foods, swallowing rehabilitation, and nutritional support by the nonoral route. It is important to evaluate the respiratory function in neuromuscular diseases leading to dysphagia, because many of these disorders disrupting deglutition may interfere with either diaphragmatic or intercostal muscle function.
When diagnosticians approach neuromuscular disorders leading to dysphagia, it is useful to begin at the muscular end of the nervous system, because a greater number of etiologies belong to this category.
Primary Muscle Diseases
Although in theory any muscular disorder may present with impairment of swallowing, abnormalities of deglutition tend to predominate in some types of muscle disease. These include certain muscular dystrophies such as oculopharyngeal muscular dystrophy (OPMD), myotonic dystrophy (MD), and rare patients in the advanced stages of Duchenne muscular dystrophy (DMD). Inflammatory disorders such as polymyositis (PM), dermatomyositis (DM), and inclusion body myositis (IBM) can involve the muscles of deglutition. Certain metabolic myopathies, particularly mitochondrial myopathies, may present with impairment of swallowing.
The patients affected with myopathy and dysphagia often present with subjective complaints of either choking on solids, or plain inability to swallow the food. Occasionally, patients comment on the fact that they cough frequently, particularly when drinking liquids. They may point to the upper cervical region or to the mid-sternal region depending on whether the esophagus is involved or not.
In a subset of myopathic patients, the dysphagia is associated with impairment of the cricopharyngeal muscle function and its ability to relax, which then prevents the food from leaving the hypopharynx into the esophagus. This is termed cricopharyngeal achalasia. The patient may describe tightness in the throat, and may complain of pooling of secretions as well as develop insidious weight loss. Radiologic evaluation of the pharynx during barium swallow often shows this appearance of cricopharyngeal achalasia. This problem can occasionally be treated with a cricopharyngeal myotomy, and a muscle biopsy specimen from the cricopharyngeus sometimes can illustrate the diagnoses, particularly in the case of inflammatory myopathy, inclusion-body myositis, or oculopharyngeal muscular dystrophy (Figures 1 and 2).
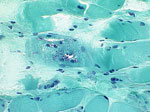
Figure 2: Cryostat sections.
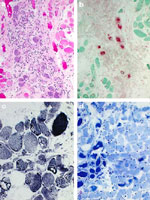
a: Granulomatous formation (hematoxylin and eosin,  200). b: Inflammatory cells (acid phosphatase, 200). c: Lobulated fibers (nicotinamide adenine dinucleotide phosphate oxidative stain, 200). d: Fiber grouping (adenosine triphosphatase, 200).
200). b: Inflammatory cells (acid phosphatase, 200). c: Lobulated fibers (nicotinamide adenine dinucleotide phosphate oxidative stain, 200). d: Fiber grouping (adenosine triphosphatase, 200).
In OPMD, patients usually develop symptoms between the fourth and fifth decades. The onset is often with ptosis, but dysphagia is frequent and may be the presenting symptom. It is usually progressive. Weakness of extraocular muscles occurs later, but diplopia or total ophthalmoplegia are rare. The dysphagia is mainly pharyngeal, but the lingual and oral phases are also affected. Proximal muscle weakness ensues, and is frequently worse in the lower limbs. After years of progression, patients develop dysphonia and aspiration. Unless repeated aspiration occurs, the life span is usually normal, although rare patients may develop cardiac conduction block. The creatine kinase (CK) level may be elevated but seldom exceeds 500 U/L. A muscle biopsy obtained from a weak muscle reveals rimmed vacuoles on routine histochemical studies. Electron microscopy shows intranuclear inclusions made of tubular filaments measuring 8.5 nm in diameter.2
Genetically, OPMD is caused by expansion of a GCG triplet repeat sequence located within the polyadenylate binding protein nuclear 2 gene (PABP2) on chromosome 14q11.2-13. Normally, six GCG repeats, coding for alanine, are expressed. Expansion of this sequence beyond eight repeats results in autosomal dominant OPMD. Patients homozygous for this expansion have the most severe phenotype, with onset of dysphagia occurring a decade or two earlier than in heterozygotes.3 Another severe phenotype occurs in patients with an expansion of the repeat sequence on one chromosome and a seven-repeat polymorphism on the other. On the other hand, patients with two copies of the polymorphic allele manifest a more benign phenotype, and in these patients limb weakness is usually mild and dysphagia may be the only presenting manifestation. The PABP2 protein is expressed in the nuclei of skeletal muscle. The expansion of the GCG repeat sequence causes abnormal lengthening and misfolding of the PABP polyalanine tail, leading to its accumulation within the intranuclear inclusions.4 Because these aggregates are resistant to degradation, their accumulation causes toxicity to muscle cells.
Myotonic dystrophy (MD, Steinert's disease) is the most common adult-onset form of muscular dystrophy. It is also the most common inherited distal myopathy. In the United States it has an incidence of 13.5 per 100,000 live births. Myotonia is an important clue to this diagnosis. Myotonia is the phenomenon of impaired relaxation of muscle after forceful voluntary contraction and is due to repetitive depolarization of the muscle membrane. Patients may complain of muscle stiffness or tightness, resulting in difficulty releasing their handgrip after a handshake, unscrewing a bottle top, or opening their eyelids if they shut their eyes forcefully. Myotonia classically improves with repeated exercise, and worsens with exposure to cold. It most commonly involves the hands, tongue, and eyelids, and can be demonstrated at the bedside by percussion of the thenar eminence, wrist extensors, or tongue. Electromyography reveals myotonic discharges with their characteristic "dive-bomber" sound. Despite its importance, patients seldom complain of myotonia, and instead complain of distal weakness. In the arms, the hand intrinsic muscles and the extensors of the fingers and wrist are preferentially affected; in the legs, patients may develop foot drop. Proximal limbs are less affected. There is weakness of the facial muscles, and atrophy of the temporalis, masseter, and sternocleidomastoid muscles, leading to a characteristic hatchet-face appearance. Ptosis and frontal balding also occur. There is frequent dysarthria and dysphagia owing to weakness of the palatal and pharyngeal muscles. The dysphagia is worsened by involvement of the smooth musculature of the esophagus. Radiologic evaluation of swallowing shows impairment of oral, pharyngeal, and esophageal phases. Swallow onset is delayed, and bolus transit time is slowed. Many patients have incomplete upper esophageal sphincter (UES) relaxation. Involvement of other smooth muscles (gallbladder, intestines) leads to other gastrointestinal (GI) complications (gallstones and intestinal pseudo-obstruction). Cardiac involvement is common and is a major source of mortality. Baseline electrocardiography is abnormal in 65%, and Holter monitoring is abnormal in 29% of patients.5 First-degree atrioventricular block is the most common abnormality. Cardiac histopathology shows fibrosis, primarily in the conducting system and sinoatrial node, myocyte hypertrophy, and fatty infiltration. Other common problems are bilateral posterior subcapsular cataracts, insulin resistance, testicular atrophy, and uterine hypotonia. Lower IQ occurs particularly with earlier age of symptom onset. Magnetic resonance imaging (MRI) of the brain can reveal hyperintense white matter lesions or cortical atrophy.6
The CK level is normal or mildly elevated. When muscle biopsy is performed, type I fiber atrophy, type II hypertrophy, sarcoplasmic masses, and ring fibers are the most common findings. A significant increase in the number of myofibers containing internal nuclei should alert to the diagnosis. Genetic testing has lessened the need for muscle biopsy. Myotonic dystrophy is an autosomal dominant disease. Patients have an abnormal expansion of a CTG trinucleotide repeat sequence located on chromosome 19q13.2.7 There is genetic anticipation, with an increase of the triplet repeat size and worsening of the phenotype in subsequent generations. The repeat sequence lies in the myotonin protein kinase (DMPK) gene. Normal controls have fewer than 35 repeats. There is some correlation between the number of repeats and the phenotype, and swallowing impairment tends to increase along with the number of repeats. Infants with congenital MD are born to an affected mother and generally have over 1000 repeats. They often suffer from neonatal hypotonia and may require mechanical ventilation because of respiratory distress. Bifacial weakness and feeding difficulties are prominent. Examination of the mother is helpful for the diagnosis, which is confirmed by genetic testing. Once children survive the infantile period, progressive muscle weakness appears later, and up to one half are mentally retarded.
In patients with advanced DMD, there is some involvement of the oropharyngeal muscles. Duchenne muscular dystrophy is transmitted as an X-linked inheritance, with female carriers and affected males. The incidence of DMD is approximately 1:3500 live births. Patients are normal at birth, but childhood motor milestones become somewhat delayed. By age 5 years, boys experience difficulty running, climbing stairs, or arising from the floor (Gowers' sign). Calf hypertrophy is an early sign. The later occurrence of macroglossia further complicates the oral phase of swallowing. There are also reports of impaired gastric motility owing to the involvement of smooth muscles. This may lead in advanced stages to esophageal dysmotility, gastric dilatation and intestinal pseudo-obstruction.8 The CK levels are markedly elevated. The average IQ is one standard deviation below the mean. Boys often become wheelchair-bound before 12 years of age. Scoliosis complicates the respiratory dysfunction in these patients. Cardiomyopathy occurs in the late teenage years, and is a frequent source morbidity and mortality. Most often, it is of the dilated type, but hypertrophic cardiomyopathy may also occur. Patients die before their third decade. The condition is caused by mutations in the dystrophin gene, located on the short arm of the X chromosome (Xp21.2). This gene codes for dystrophin, a large intracellular protein that links the intracellular cytoskeleton and extracellular matrix and provides stability to the muscle membrane.
The primary inflammatory muscle diseases are the largest group of acquired and potentially treatable myopathies. They are differentiated into three main subsets: PM, DM, and IBM. Dermatomyositis is a microangiopathy affecting skin and muscle; there are perivascular B and CD4 lymphocyte infiltrates, and there is activation and deposition of complement, which causes lysis of endomysial capillaries and muscle ischemia. In PM and IBM, CD8+ cytotoxic T cells invade muscle fibers that express major histocompatibility complex (MHC) class I antigens, which leads to fiber necrosis mainly via the perforin pathway. In IBM, there is formation of vacuoles with amyloid deposits as well. The responsible autoantigen(s) has not yet been identified. The upregulation of various cell adhesion molecules and cytokines contribute to the immunopathologic process. Early diagnosis and initiation of therapy is essential, because both PM and DM respond to immunosuppressive agents. Inclusion body myositis tends to be less responsive to treatment.9
Clinically, PM and DM are characterized by a proximal weakness that develops in weeks to months. The CK levels are often elevated. Cutaneous involvement is specific to DM, which may have infantile or adult onset. Polymyositis and IBM are disorders of adults. Swallowing disorders may be severe in these patients, and complicate significantly any respiratory dysfunction they may have (aspiration, interstitial lung disease, respiratory muscle deficiency). Electrodiagnostic testing (electromyography, EMG) and muscle biopsy are particularly important for the diagnosis. In certain patients, the detection of specific myositis autoantibodies provides further clues. The most common antibodies are antisynthetase or anti-Jo1 in PM/DM with interstitial lung disease, and anti-Mi-1 and -2 in DM. Adult PM and DM may be associated with connective-tissue disease (overlap syndrome) or cancers. Some cases of PM are secondary to infections (human immunodeficiency virus, human T-cell leukemia virus-1, and toxoplasmosis). Inclusion body myositis is the most frequent acquired myopathy after 50 years of age; it is characterized by early distal muscle weakness, slow course, and somewhat poor response to corticosteroid and immunosuppressive agents. Histologically, there are rimmed vacuoles and filamentous inclusions in addition to the cytotoxic inflammatory process. On immunofluorescence,  -secretases that cleave amyloid- precursor protein colocalize with amyloid- in IBM vacuolated muscle fibers.10
-secretases that cleave amyloid- precursor protein colocalize with amyloid- in IBM vacuolated muscle fibers.10
In inflammatory myopathies, dysphagia occurs primarily from the involvement of striated muscles, but in some cases the upper third of the esophagus may be affected. This is particularly true in overlap PM seen with systemic sclerosis or mixed connective tissue disorders. In one study of 62 patients with systemic sclerosis or related disorders referred for evaluation of upper GI symptoms, dysphagia was present in 61% of patients. Other GI symptoms were heartburn (77%), nausea/vomiting (58%), diarrhea (53%), constipation (31%), and fecal incontinence (13%). Manometric studies in 36 patients showed antral hypomotility and reduced amplitude and frequency of intestinal contractions.11 In addition to the dysphagia, patients may have deglutitive pharyngeal and laryngeal pain, and may develop aspiration of their food.
Dysphagia may be an early feature in older patients affected with IBM.12 Indeed, dysphagia may be the presenting symptom in older individuals with inflammatory myopathy. It can be isolated. Fiberoptic laryngoscopy reveals pooling of saliva in the pharyngeal recesses. The swallowing videofluoroscopy shows a prominent cricopharyngeus muscle, and some patients have a prominence in a more proximal portion of the inferior constrictor muscle. Biopsies of the cricopharyngeus muscle may show the inflammatory changes.13
Previous retrospective series suggested that PM is the most common type of myositis, followed by DM and the overlap syndromes in which PM or DM is associated with connective tissue disorder. However, idiopathic PM is probably less common than other types of inflammatory myopathy, such as IBM, DM, or overlap PM, particularly when dysphagia is prominent.14 In a recent longitudinal study of 100 consecutive adult French-Canadian patients with inflammatory myopathies, patients were reclassified based on their clinical, laboratory (autoantibodies), and biopsy data. At presentation, PM was the most common diagnosis, accounting for 45% of the cohort. After complete workup, its frequency fell to 14%. Conversely, the frequency of myositis associated with connective tissue disease increased from 24% to 60%.15 In that series, systemic sclerosis was the most common connective tissue disease associated with PM, accounting for 29% of the cohort. This is useful in guiding immunosuppressive therapy, because DM and overlap PM appear to respond to therapy better than PM or IBM.
In certain mitochondrial myopathies, such as Kearns-Sayre, chronic progressive external ophthalmoplegia, and MNGIE syndrome (mitochondrial myopathy, peripheral neuropathy, gastrointestinal disease, and encephalopathy), the patients present with dysphagia owing to the primary involvement of the pharyngeal constrictor muscles, but also with weakness of the oropharyngeal musculature. In one study of 12 patients, cricopharyngeal achalasia was present in nine, and deglutitive incoordination was found in one patient. In MNGIE syndrome, there is additional smooth muscle involvement, which may result in intestinal pseudo-obstruction owing to the visceral neuropathy.16
Dysphagia is rather uncommon in congenital myopathies. Among these, nemaline rod myopathy is probably the one most associated with impaired deglutition. Patients usually present with severe neonatal hypotonia. When respiratory distress is present, death can occur in the first year of life. There is delay of motor milestones during childhood. Patients have significant facial weakness, a high arched palate, micrognathia, and weak masseter and pterygoid muscles. The pharyngeal and laryngeal muscles may be affected, but ptosis and extraocular muscle involvement do not occur. Muscle stretch reflexes are absent. Skeletal anomalies such as scoliosis, pectus excavatum, clubfoot, and pes cavus are common. Dysphagia does not occur in the adult form, which presents with a limb-girdle phenotype. The CK levels are normal, but the EMG shows myopathic changes. Muscle biopsy shows rods (red clusters) in the subsarcolemmal zone. Ultrastructurally, the rods arise from the Z disk. Inheritance is usually autosomal dominant, rarely autosomal recessive. To date, mutations have affected genes encoding for various skeletal proteins, such as tropomyosin-3, actin, and nebulin.17
Neuromuscular Junction Disorders
Disorders of the neuromuscular junction also lead to dysphagia. Myasthenia gravis (MG) characteristically presents with intermittent swallowing difficulties that worsen throughout the meal or throughout the day, and are often associated with oculomotor abnormalities, diplopia, and facial muscle weakness. Fatigable muscle weakness is clinically the hallmark of this disease. Dysphagia in MG is due to disordered oral and pharyngeal phases, which prospective videofluoroscopic studies have clearly shown. In a small number of older patients, manometric studies have shown weak peristaltic contractions of the esophagus.18 Clinically, some patients have greater problems with chewing food or moving it in their mouth, whereas others have difficulties restricted to the pharyngeal phase. Several of these patients also have a nasal speech owing to weakness of the soft palate, whereas other dysphagic patients may have dysphonia. About one third of dysphagic patients aspirate.19, 20 Dynamic EMG studies show greater impairment in the submental and laryngeal muscles, with relative sparing of the cricopharyngeal muscle.21 When dysphagia occurs in elderly patients with MG, it tends to be less responsive to immunomodulatory treatment, such as plasmapheresis or IV gamma globulins.22 The presence of dysphagia also increases the risk of respiratory failure, hence the necessity of therapeutic urgency. Involvement of limb musculature may follow, but the abnormalities may remain confined to cranial and bulbar musculature. These patients should have the usual workup for myasthenia, including evaluation of thyroid function, measurement of acetylcholine receptor antibodies, anti-MuSK (muscle-specific receptor tyrosine kinase) antibodies, and single-fiber EMG.23 A thorough search for a thymic abnormality should be done, and it should be treated similarly to any generalized myasthenia even when limb weakness does not appear significant. Treatment includes cholinesterase inhibitors, corticosteroids, and plasmapheresis with occasional use of intravenous gamma globulins.24
Involvement of the pharyngeal muscles in Lambert-Eaton syndrome has been described, but it is not common. Only 7% of patients develop dysphagia.25 Additional workup for a neuromuscular transmission defect often reveals immunologic changes such as the presence of calcium voltage-gated channels owing to either disimmune disorder or a latent small cell lung cancer.26 The treatment entails addressing the underlying etiology, and prescribing 3,4-diaminopyridine and plasmapheresis if necessary.
Botulism presents selectively with weakness of swallowing that spreads into other muscle groups. Traditional cases are associated with poorly preserved food, but more recent cases have been seen as a complication of injections with botulinum toxin A or B to treat various cervical and bulbar dystonias.
Peripheral Nerve and Motor Neuron Disorders
Peripheral neuropathy seldom involves the pharyngeal muscles, given the short length of the pharyngeal nerve fibers. However, disorders that are independent of fiber nerve length tend to affect the bulbar muscles. This can be seen in polyradiculoneuropathies, both acute (Guillain-Barré syndrome) and chronic (chronic inflammatory demyelinating polyradiculoneuropathy), often in the setting of other cranial nerve involvement.27, 28 In rare cases of Guillain-Barré syndrome, the pharyngeal and cervical muscles are predominantly affected, and these patients have prominent dysphagia. Certain etiologies of acute polyradiculoneuropathy such as diphtheria present selectively with weakness of swallowing followed by the usual picture of weak areflexic limbs with distal sensory loss. In chronic inflammatory demyelinating polyradiculoneuropathy, bulbar muscles may become affected and dysphagia occurs in about 16% of patients; it usually involves the pharyngeal phase, but oral manipulation mainly for solid food is present in dysarthric patients. The treatment entails preventing other complications such as aspiration, and addressing the etiology more specifically. Acute inflammatory demyelinating polyneuropathy is treated by the best medical care, supplemented by the use of intravenous gamma globulin or plasmapheresis. In chronic inflammatory neuropathies, these modalities often are used in conjunction with corticosteroids.
Patients with motor neuron disorders can present with dysphagia. Among hereditary chronic motor neuron diseases, dysphagia tends to occur early in bulbospinal muscular atrophy (Kennedy's disease). This is an X-linked disorder in which there is a triple CAG repeat on the long arm of the X chromosome, which leads to degeneration of bulbar motor neurons and reduction in tissue responsiveness to androgen. A small percentage of these patients also develop mild polyneuropathy as well as glucose intolerance. The syndrome should be suspected in a middle-aged man who presents with dysphagia and gynecomastia. Once the diagnosis is confirmed by genetic testing, conservative support is necessary. One should also consider genetic counseling of first-degree relatives.29
A more ominous disorder that involves bulbar muscles is ALS, also known as Lou Gehrig's disease. In this condition, there is an accelerated death of motor neurons, which is often asymmetric at least in the early stages. When bulbar motor neurons become affected, this leads to weakness of the orolingual as well as the pharyngeal muscles. These patients develop swallowing difficulties particularly to solids, but given the frequent presence of an upper motor neuron dysfunction, they also sometimes choke on fluids or have frequent regurgitation. Bulbar involvement occurs in up to 20% of the patients after 1 year of diagnosis, and the percentage increases steadily with further progression. Serial video-manometric studies show a decrease in the swallowing pressure first in the oropharynx and then in the hypopharynx, but most patients retain their UES relaxation.30 However, EMG studies of the cricopharyngeal muscle show delayed UES opening or premature UES closing.31 These patients often lose weight in part because of dysphagia, but also because there is denervation of limb muscles and significant reduction of appendicular muscle mass. Complications such as aspiration pneumonia become rather eminent. Several of these patients should be counseled regarding the insertion of a gastric feeding tube. Once dysphagia becomes prominent, respiratory difficulties often follow soon, and the disorder becomes fatal.
Other chronic motor neuron disorders such as spinal muscular atrophy sometimes affect the muscles of swallowing, except in the infantile variant (type 1) known as Werdnig-Hoffman disease. These children are often born hypotonic and floppy with weak suck and limb motion. Most children die in the first 3 years of life.32
Conclusion
Table 1 summarizes the main features of dysphagia in various neuromuscular disorders. Once dysphagia becomes overt, it is important to address the more specific management of the underlying etiology. In addition to the social incapacitation induced by impaired deglutition, patients often suffer from impaired nutrition and are at a higher risk of aspiration of fluid. Workup would confirm the involvement of swallowing muscles, and reveal the presence of pooling and aspiration. The presence of UES impairment can also be diagnosed. Laryngeal and pharyngeal EMG33 confirm the presence of an underlying neuromuscular condition, and indicate the need for a more selective workup, including in some cases a muscle biopsy.34 Adequate management can then be pursued. If cricopharyngeal achalasia is found, UES dilatation or focal injection of botulinum toxin may be helpful. When Zenker's diverticulum or cricopharyngeal achalasia is found, surgical correction is necessary. In selected cases, cricopharyngeal myotomy is usually successful in alleviating the dysphagia for several years.35 It is also important to evaluate the esophageal phase of swallowing, because involvement of the smooth musculature is helpful in further clarifying the etiologic diagnosis, and would alter the profile of management. In patients who respond poorly to the various treatments, feeding tubes may be required.

