
Abstract
IFN alfacon-1 (Infergen) is a synthetic form of Interferon (IFN)-α2b. Infergen has immunomodulatory activity and is effective against hepatitis C virus. However, the effect of Infergen (IFG) on Mycobacterium tuberculosis (Mtb) has not yet been reported. Therefore, for the first time, we have studied the influence of IFG in constraining the survival of Mtb in human macrophages. We observed that IFG significantly enhanced the maturation and activation of macrophages. Further, it substantially augmented the secretion of IL-6, nitric oxide (NO) and antigen uptake. Moreover, macrophages exhibited remarkably higher bactericidal activity, as evidenced by reduction in the Mtb growth. Infergen-mediated mechanism was different from the type-1 interferons; since it worked through the activation of NF-κB, phosphorylation of STAT-3 and Akt-PI3K that improved the bactericidal activity through autophagy and NO release. In future, IFG immunotherapy can be a novel strategy for treating patients and controlling TB.
Similar content being viewed by others


Introduction
In spite of the fact that effective drugs like rifampicin, isoniazid, ethambutol, pyrazinamide, etc., are available to treat tuberculosis (TB); the disease continues to inflict a detrimental impact on public health worldwide. Nearly one third of the world population is infected with Mycobacterium tuberculosis (Mtb) and about 1% develops new infection annually, accounting for total 9.6 million new cases1. In 2014, there were about 1.5 million allied deaths; typically occurring in the developing countries [WHO, 2015]. The problem is further aggravated with the emergence of MDR-TB (multi-drug-resistant tuberculosis), XDR (extensively drug-resistant tuberculosis) and TDR (totally drug-resistant tuberculosis) strains of Mtb, AIDS-pandemic and failure of BCG vaccine2,3,4. Hence, there is an urgent need to identify newer drugs and develop radical changes in the current therapeutic strategies to control TB.
Interferons (IFNs) are naturally occurring immunomodulatory cytokines. Type-I IFNs are very well-known for their anti-viral activity5,6,7,8,9. Recently, a non-naturally occurring type-I IFN known as IFN alfacon-1 or Infergen (IFG) has been approved against hepatitis C virus10,11. Infergen is quite distinct from type-I IFNs, since it is derived from the sequences selected from several α-IFNs (interferon α2a and interferon α2b) and then producing a consensus gene product12. The role of type-1 IFNs against Mtb has not been clear so far13,14,15,16. However like IFN-γ, type-1 IFNs are reported to kill Mtb by bolstering host immunity13,14,15,16.
Although, IFG shares biological and pharmacological properties with type 1 IFNs, it follows a unique and distinct signaling pathway. Infergen exhibits potent immunomodulatory activity in human cells17. Furthermore, it is known to control viral infection by enhancing the activation of T cells through an augmented viral peptide presentation by antigen presenting cells (APCs). Unfortunately, nothing has been reported regarding the therapeutic aspect of IFG on Mtb. Therefore, we thought that it would be interesting to study the impact of IFG in restricting the intracellular survival of Mtb and the mechanism involved therein.
Autophagy plays an essential role in protection against Mtb18. It targets the Mtb antigens to lysosomes for their degradation and clearance. Further, autophagy enhances the antigen presenting ability of APCs to T cells19,20. At the same time, it inhibits detrimental inflammatory responses21. Nitric oxide (NO) is known to confine the growth of Mtb22. Taking into account these evidences, we were interested to monitor the influence of IFG on the activation of Mtb infected macrophages, thereby restricting the survival of the Mtb. It was quite fascinating to observe that IFG stimulated macrophages displayed significantly improved skill to restrict the intracellular growth of Mtb. The mechanism involved was through the augmentation of autophagy and release of NO. Both these pathways are considered crucial in curbing the growth of Mtb. Therefore, this study illustrates that IFG may have enough potential to work as an immunotherapeutic agent against TB.
Results
IFG restricts the intracellular survival of Mtb
Mtb not only infects macrophages but also it can survive in the hostile environment of these cells23,24. Thus, in the initial phase of the study, we investigated the impact of IFG on avirulent strain of Mtb H37Ra infected THP-1 macrophages (H37Ra-Mφ). It was quite exciting to note that IFG stimulated macrophages (MφIFG) showed significant (p < 0.0001) reduction in the intracellular survival of Mycobacterium, as compared to unstimulated control [Fig. 1A]. The decrease in the CFUs was observed in a dose dependent fashion. Likewise, substantial (p < 0.0008) decline in the growth of virulent strain H37Rv (Mtb) was noticed [Fig. 1B]. Furthermore, we proved the above event through time-point kinetics by demonstrating a extensive (p < 0.0001) increase in Mtb killing (GFP-H37Ra) by flow cytometry assay [Fig. 1C]. Thus, validating the potent role of IFG in inhibiting the growth of both virulent and avirulent Mtb. The concentration of 64 ng/ml of IFG showed the maximum intracellular killing of Mtb and was not toxic to Mφ and human peripheral blood mononuclear cells (PBMCs) [Fig. 1A and Fig. S1A–C]. Hence, this dose was chosen in all the subsequent experiments.

IFG stimulated macrophages show augmented killing of Mtb.
Macrophages were infected for 4 h with the mycobacterial strains [A] H37Ra; [B] H37Rv. Later, cells were stimulated with the indicated doses of IFG for 24 h. The infected macrophages were lysed and CFUs were enumerated after 21d. Infected macrophages treated with INH (25 μg/ml) were used as positive control. [C] Time-point kinetics of GFP-H37Ra after 4 h infection. This was followed by stimulation with IFG for 24 h and 48 h. The flow cytometry data (iMFI) represented through the line diagrams as mean ± SEM are representative of two independent experiments. [D] Supernatants of the H37Ra infected macrophages stimulated with IFG were collected after 24 h for the estimation of IL-6 by ELISA; [E] RNA was isolated after 6 h to monitor the expression of IL-6 by RT-qPCR. Data depicted as bar diagram are mean ± SEM of 4 individual wells and representative of four independent experiments. **p < 0.001, ***p < 0.0001. [F,G] Macrophages isolated from human PBMCs were infected with Mtb for 4 h. Later, they were stimulated with IFG for 24 h and the supernatants were collected for the estimation of IFN-γ and IL-6. UI: uninfected control; US: Mtb infected macrophages; IFG: Mtb infected and IFG (64 ng/ml) stimulated macrophages; INH: Mtb infected macrophages treated with isoniazid. Data represented as the mean ± SEM are of four wells and two independent experiments. **p ≤ 0.006, ***p ≤ 0.0006, ****p < 0.0001.
IFG augments the secretion of cytokines
IL-6 is a major cytokine produced by macrophages in response to intracellular pathogens25. This cytokine plays an important role in T cell activation and inhibition of the growth of Mtb25,26,27,28,29,30,31. IFG stimulated H37Ra infected Mφ produced considerably (p < 0.001) higher level of IL-6, as compared to unstimulated (no Infergen) macrophages infected with H37Ra [Fig. 1D]. The increase in IL-6 was observed in a dose dependent manner. Further, these results were corroborated by IL-6 gene expression by RT-qPCR [Fig. 1E]. Additionally, we confirmed these results by observing a significant (p < 0.01) increase in the level of IL-6 and IFN-γ by IFG stimulated H37Ra infected macrophages isolated from human PBMCs [Fig. 1F,G]. Infergen is reported to promote Th1 polarization32. No noticeable change was seen in IL-1β, TGF-β, IL-10 and TNF-α genes expression [Fig. S2A–D].
IFG upregulates the expression of CD80, CD86 and HLA-DR
It has been reported that signaling through IFN-α/β can activate macrophages and other cells of the immune system during viral infection31,32,33,34,35. Further, MHC and costimulatory molecules expressed on the surface of the macrophages are crucial for the optimum activation of T cells. It was noticed that Mtb-infected IFG stimulated THP-1 macrophages (Mtb-Mφ-IFG) exhibited substantially (p < 0.02) higher expression of costimulatory molecules CD40 (p < 0.0004), CD80 (p < 0.0001), CD86 (p < 0.02) and MHC-II (HLA-DR) (p < 0.003) [Fig. 2A–D and Fig. S3A–D]. These results were substantiated using CD11b+ human macrophages. We observed that IFG induces significant upregulation of CD80 (p < 0.01), CD86 (p < 0.05), compared to unstimulated controls [Fig. 2E,F and Fig. S3E,F]. Thus, our data suggest that IFG not only augments the activation and maturation of Mtb infected Mφ differentiated from THP-1, but also macrophages isolated from human peripheral blood.

IFG upregulates the expression of CD40, CD80, CD86, and HLA-DR on the Mtb infected macrophages.
[A–D] Macrophages; [E,F] Human PBMCs were infected with Mtb for 4 h and subsequently stimulated with IFG (64 ng/ml) for 24 h. Later, expression of CD40, CD80, CD86, and HLA-DR was evaluated by flow cytometry on the [A–D] macrophages; [E,F] CD11b gated PBMCs macrophages. The flow cytometry data (iMFI) represented through bar diagrams as mean ± SEM are representative of two independent experiments. UI: uninfected macrophages; UI + IFG: uninfected macrophages stimulated with IFG; US: Mtb infected macrophages; Infected + IFG: Mtb infected and IFG stimulated macrophages. *p ≤ 0.0285, **p < 0.003, ***p < 0.0004, ****p < 0.0001.
We further checked the activation status of other immune cells like B cells and T cells. As compared to untreated B cells, IFG treated B cells displayed a higher expression of CD80, CD86 and HLA-DR, [Fig. S4A–F]. We also noted significant (p < 0.01) upregulation of the activation marker CD25 on CD8 T cells treated with IFG [Fig. S5A]. Moreover, IFG incubated CD8 T cells showed substantial improvement (p < 0.01) in their ability to lyse the target cells [Fig. S5B]. Furthermore, IFG induced significant (p < 0.05) proliferation of PHA elicited human PBMCs [Fig. S5C]. These results designate that IFG not only stimulates the macrophages, but also B cells and CD8 T cells.
IFG activated macrophages show enhancement in antigen uptake
Macrophages are extremely efficient APCs responsible for phagocytosis. Therefore, we were curious to check whether IFG stimulated macrophages acquire an enhanced capacity to phagocytose Mtb. Consequently, we stimulated macrophages with IFG and then infected them with Mtb. The extracellular bacteria were removed by extensive washings and subsequently treated with amikacin. Later, the cells were lysed and bacterial growth was enumerated by CFU plating. Remarkably, IFG significantly (p < 0.05) improved the ability of macrophages to phagocytose H37Ra, compared to unstimulated control [Fig. 3A]. These results were further supported by increased uptake of other mycobacterial strains M. smegmatis [Fig. 3B] and Mtb-H37Rv [Fig. 3C]. Furthermore, our flow cytometry and confocal microscopy experiments substantiated higher uptake of GFP+-H37Ra by IFG stimulated Mφ [Fig. 3D,E and Fig. S6A,B]. The Z-stack [Fig. S6C] and live imaging videos [Fig. S6D, video 1, 2] also confirmed the presence of GFP+-Mtb inside the macrophages. In addition, we validated the higher uptake capacity of GFP+-H37Ra by IFG stimulated Mφ through time point kinetics assay by flow cytometry [Fig. 3F]. Thus, these results signify the potential role of IFG in enhancing the phagocytosis by macrophages.

Signaling through IFG enhances phagocytosis.
Macrophages stimulated with IFG for 24 h were infected with [A] H37Ra; [B] M. smegmatis; [C] H37Rv; [D,E] GFP-H37Ra. After 4 h, cells were lysed and bacterial uptake was monitored by [A–C] CFUs. [D] Flow cytometry; [E] confocal microscopy. [A–C] The data (mean ± SEM) shown as bar diagrams indicate bacterial uptake. [D] The data in the inset of histograms shown as % cells/MFI denote bacterial uptake. [F] The macrophages stimulated with IFG for 24 h and thereafter infected with GFP-H37Ra for 4 h. Time-point kinetics of bacterial uptake by macrophages was measured by flow cytometry. The flow cytometry data (iMFI) represented through the line diagrams as mean ± SEM are representative of two independent experiments. IFG: infected macrophages stimulated with Infergen; US: infected macrophages (not stimulated with IFG); DIC: differential interference contrast. Image magnification: 60x. Data are representative of two independent experiments. *p < 0.05, **p < 0.003, ***p < 0.0004, ****p < 0.0001.
Induction of iNOS expression by IFG is necessary for autophagy
We next monitored the mechanism of action operating in IFG stimulated cells in restricting Mtb survival. Intriguingly, we observed that inhibition in the growth of Mtb by IFG stimulated cells was operating through NO production, as evidenced by an increase in the expression of INOS by RT-qPCR [Fig. 4A], as well as through expression of iNOS in the cytosol by Western blotting experiments [Fig. 4B]. Further, Mtb infected Mφ on stimulation with IFG produced remarkably more nitric oxide (NO), as compared to either unstimulated or infected control macrophages [Fig. 4C]. Nitric oxide is an established indicator of bactericidal activity36. We substantiated the specificity of the induction of iNOS by treating Mtb infected macrophages with iNOS inhibitor [(N-monomethyl-L-arginine), NM]. Interestingly, we observed significant decline in the NO production (p < 0.0001) and increase in the bacterial CFUs (p < 0.05) upon treatment of Mtb-MφIFG with NM [Fig. 4C,D]. We also showed that there was downregulation in the autophagy marker LC3, when iNOS was inhibited by NM [Fig. 4E]. Thus, establishing that iNOS activity is required for autophagy. Furthermore, autophagy inhibitor 3 methyl adenine (3MA) was used to unambiguously establish the induction of autophagy in Mtb infected Mφ treated with IFG [Fig. 4E]. Rapamycin was used as a positive control. Moreover, blocking of Akt-PI3K and NF-кB pathways by their respective inhibitors, substantially reduced the secretion of NO [Fig. 4F].

IFG restricts the intracellular survival of Mtb through the induction of iNOS.
Macrophages were infected with Mtb for 4 h. Later, cells were stimulated with IFG (64 ng/ml) for 24 h and monitored for the expression of [A] INOS gene by RT-qPCR; [B] iNOS by Western blotting and values [fold change] are shown through densitometry analysis. [C,D] The specificity of NO production and its role in Mtb killing is proven by NO inhibitor NM. The estimation of [C] NO was done by Griess method and [D] Mtb killing by CFUs. The impact of autophagy in the inhibition of the growth of Mtb was ascertained by the [E] conversion of LC3-I to LC3-II by Western blotting and densitometry values are shown as fold change. Actin was used as a loading control. [F]. The secretion of NO was inhibited by treating the cells with the inhibitors of Akt and NF-κB. LPS was used as a positive control. The specificity of autophagy was ascertained by blocking the conversion of LC3-1 into LC3-II by its inhibitors 3MA and NM. Rapamycin is used as a positive control of autophagy. The fold change was calculated considering the value of infected controls as 1. The data are representative of 2–4 independent experiments. UI: uninfected macrophages; Infected: infected macrophages and unstimulated (no IFG); IFG: Mtb infected macrophages and IFG stimulated, NM: N-monomethyl-L-arginine, 3MA: 3 methyl adenine. *p < 0.05, **p < 0.01, ****p < 0.0001.
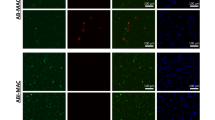
Autophagy plays a fundamental role in inhibiting the intracellular survival of Mtb37. Interestingly, IFG stimulation exhibited higher conversion of LC3I to LC3II, a hallmark of autophagy. We observed further amplification of autophagy by rapamycin treatment of the infected macrophages stimulated with IFG [Fig. 5A]. Thus corroborating the IFG induced autophagy. Additionally, blocking of Akt and PI3K pathways by their respective inhibitors inhibited the induction of autophagy [Fig. 5B]. Furthermore, augmentation (p < 0.0001) of the autophagy genes ATG5, ATG7 and BECLIN1, categorically established the involvement of autophagy [Fig. 5C–E]. Finally, we verified this finding by observing the over expression of autophagy influx by LC3 puncta formation in GFP-H37Ra infected macrophages through confocal microscopy after IFG and rapamycin treatment [Fig. 5F,G]. We observed that virulent and avirulent mycobacteria have similar responses with respect to autophagy. Interestingly, we also observed that IFG itself has an ability to induce autophagy influx in macrophages (Fig. S7A,B). Likewise, the acidic vacuoles were stained with acridine orange dye to show the induction of autophagy [Fig. 5H,I]. We also monitored autophagy by treating cells (infected and IFG stimulated) with rapamycin and correlated this with NO production and CFU burden [Fig. 5J,K]. Autophagy specific inhibitors 3MA and chloroquine (CQ) were used to establish the role of autophagy in restricting the growth of Mtb by IFG stimulated Mφ. Substantial (p < 0.0001) reduction in the secretion of NO and increase in the survival of Mtb was noticed [Fig. 5L,M]. Thus, these results signify that the function of iNOS and autophagy are interlinked with each other and are possible mechanism operating in MφIFG in constraining the Mtb growth.

The mechanism involved in controlling the growth of Mtb by IFG is through autophagy.
Macrophages were infected with Mtb (H37Rv) for 4 h. Later, cells were stimulated with IFG (64 ng/ml) for 24 h and monitored for [A] the augmentation in the IFG induced autophagy by rapamycin through conversion of LC3-I to LC3-II by Western blotting. The densitometry values are shown as fold change. [B] The specificity of autophagy was determined by blocking Akt-PI3K pathway by their respective inhibitors. Rapamycin is used as a positive control of autophagy and actin as a loading control. The fold change was calculated considering the value of infected controls as 1. [C–E] Upregulation in the expression of autophagy markers [C] ATG5; [D] ATG7; [E] BECLIN 1 was done by RT-qPCR and the results are represented as relative expression (fold change). The fold change was calculated considering the value of uninfected controls as 1. [F,G] The induction and over expression of autophagy influx by LC3 puncta formation through confocal microscopy after IFG+rapamycin treatment in GFP-H37Ra infected macrophages. The cells were infected with GFP-H37Ra [green] and stained for the expression of LC3 with anti-LC3 Abs [red], along with nucleus with DAPI dye [blue]. The frequency of puncta formation was enumerated using 4–5 different microscopic fields and the percent of cells with LC3 puncta [mean ± SEM] are presented as bar diagram. [H,I] Acidic vacuole formation was performed by acridine orange staining and fluorescence microscopy. The data [mean ± SEM] displayed as percent positive vacuole forming cells [orange] are from five different microscopic fields. The specificity of autophagy was determined by blocking its function by autophagy inhibitors [J,K] 3MA and [L,M] CQ; as observed by the change in the yield of NO and survival of Mtb. Rapamycin was used as a positive control. The data are denoted as mean ± SEM for NO [μM] and H37RV [CFU/ml]. The results are representative of 2–4 independent experiments. UI: uninfected, US: infected, but not stimulated with IFG, IFG: Mtb infected and IFG stimulated, NM: N-monomethyl-L-arginine, 3MA: 3 methyl adenine, CQ: Chloroquine. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
IFG induces the phosphorylation of STAT-3 and activates Akt-PI3K pathway
It has been well established that IFN-α/β interacts with its receptor IFNAR and activates tyrosine kinase Jak-1 along with Tyk-238,39. Jak-1 stimulates and phosphorylates STAT-1/STAT-3. As a result, Akt is activated through STAT-3 along with PI3K and translocates into the nucleus. Further, it helps in delivering survival signal through the activation of NF-κB. It has been reported that like IFN-α/β, IFG exhibits affinity for IFNAR and activates tyrosine kinase Jak-1 along with Tyk-2. Therefore, we investigated the mechanism involved in IFG-mediated signalling events responsible for activating macrophages to restrict the intracellular growth of Mtb. Mtb infected macrophages were stimulated with IFG and phosphorylation of STAT-3, PI3K and Akt molecules were monitored by Western blotting in the cytosolic extract. Noticeable augmentation in the phosphorylation of STAT-3, PI3K and Akt was observed in the MφIFG [Fig. 6A–C]. The decrease in the phosphorylation of STAT-3 and PI3K was checked in Mφ-IFG by treating with their respective inhibitors [Fig. 6A,B]. Further, augmentation in the anti-apoptotic molecule, Bcl-xL was noticed [Fig. 6D]. Bcl-xL plays a vital role in providing a survival signal to the cells. No change was seen in the case of STAT-1 [Fig. 6E].

IFG signaling of the macrophages induces phosphorylation of STAT-3 through Akt-PI3K pathway and bolsters NF-κB nuclear translocation.
Mtb infected macrophages were cultured with IFG. The cell lysate (cytosolic extract) was prepared and Western blotting was performed to monitor the expression of [A] pSTAT-3; [B] pPI3K; [C] pAkt; [D] Bcl-xL; [E] pSTAT-1. [F,G] NF-κB was detected using nuclear extract by EMSA and densitometry values are represented by bar diagram as a fold change (mean ± SEM). [H] The translocation of NF-κB from cytosol [US] to nucleus [IFG] was further validated by confocal microscopy using IFG stimulated Mtb infected macrophages. The green fluorescence indicates NF-κB marker p65 and blue signifies the nucleus staining with Hoechst dye (magnification: 100x). LPS is used as a positive control. US: infected macrophages [no IFG]; IFG: infected macrophages treated with IFG. Data are representative of two independent experiments. ***p ≤ 0.0002.
IFG signaling augments the translocation of NF-κB into the nucleus
NF-κB is responsible for cellular growth, inflammatory and immune responses. We observed a significant (p ≤ 0.0002) increase in the activation of NF-κB in H37Ra-Mφ-IFG [Fig. 6F,G]. Further, experiments were performed that confirmed the translocation of NF-κB by confocal microscopy in IFG stimulated cells [Fig. 6H]. This observation substantiated EMSA data [Fig. 6F]. Overall, this finding signifies that induction of autophagy in IFG stimulated macrophages operates through NF-κB, STAT-3 and Akt-PI3K pathways [Fig. 7].

Proposed model of signaling pathway induced by IFG in the Mtb infected macrophages.
It has been reported that IFG binds to IFNAR-2 and activates the tyrosine kinase Jak-1 and Tyk-2. Jak-1 induces and stimulates the phosphorylation of STAT-3 [pSTAT-3]38,39. We investigated the role of IFG mediated signaling mechanism in activating macrophages to restrict the intracellular growth of Mtb. [1] The phosphorylation of STAT-3 is induced by Jak-1. [2] Subsequently, Akt is activated through STAT-3 along with PI3K. [3,4] This signaling translocates NF-κB into the nucleus and upregulates the IL-6 secretion, which receives a feed-back mechanism through pSTAT-3, Akt and therefore induces autophagy. Thus, ensuring the activation and survival of the Mtb infected macrophages. [5] The induction of autophagy in the Mtb infected macrophages directly involved in the killing of Mtb. [6] We propose that pSTAT-3 binds to the promoter sequence of INOS gene to produce ions in the cytosol that generates NO, a potent antimicrobial agent that may ultimately restrict the growth of Mtb. The data were validated by blocking the Akt-PI3K, iNOS and autophagy with their respective inhibitors to establish the role of autophagy in restricting the growth of Mtb by IFG stimulated macrophages. [7] Blocking of the Akt-PI3K, iNOS (NM), autophagy (3MA|CQ) with their specific inhibitors and therefore substantial reduction in the secretion of NO and autophagy, as well as increase in the survival of Mtb signifies that autophagy is directly involved in the killing of Mtb. Signifying that the function of iNOS and autophagy are interlinked. Overall, this mechanism illustrates that IFG plays a key role in controlling the survival of Mtb inside macrophages.
Discussion
There is an urgent need and a challenge to discover new drugs to treat TB because the disease continues to kill millions of people annually. Further, despite of the availability of potent drugs, the current regime remains quite complicated due to lengthy treatment, side effects and emergence of drug-resistant strains of Mtb.
Type 1 IFNs (IFN-α/β) are produced by macrophages, plasmacytoid DCs, fibroblasts and endothelial cells and play crucial effector function in viral infections34,40. Undeniably, previous references have indicated the efficiency of the IFNs therapy in treating yellow fever, chronic hepatitis B (HBV), hepatitis C (HCV) and ebola virus infections41,42. They augment the synthesis of several key antiviral mediators, including 2′-5′ oligoadenylate synthetase (2′-5′ A synthetase) and protein kinase R43,44. Further, type 1 IFNs upregulate CXCL10 expression in Mtb infected dendritic cells, which could be important to induce the homing and recruitment of cells to the granulomas45.
Considering the importance of IFN-α/β in activating cells of the immune system and bolstering immunity, a synthetic interferon IFG-α2b, which is known as Infergen (IFG) was synthesized46,47,48. Infergen has been reported to upregulate MHC-I expression and enhance the presentation of viral peptides by APCs. As a result, it enhances the activation and cytolytic activity of CD8+ cytotoxic T lymphocytes (CTLs)49,50,51. Further, like IFN-α/β, IFG exhibits an optimized affinity for IFNAR and augments the survival and activation of neutrophils, macrophages, DCs, NK cells, B cells and CD8 T cells31,32,33,34,35,52. Furthermore, it is reported that IFG activates various cell types to endorse viral clearance by promoting apoptosis of infected cells34,52. Unfortunately, nothing has been reported regarding the role of IFG against Mtb.
In the past, regular efforts have been made to investigate the impact of new molecules or delineate novel role of previously established molecules in the treatment of TB. Therefore, keeping in view the above-mentioned reports, we thought to evaluate the impact of IFG on the activation of macrophages and inhibition of the intracellular survival of Mtb. Following major findings have emerged from our study. IFG significantly i) inhibited the growth of Mtb in the macrophages; ii) augmented the secretion of IL-6 and IFN-γ; iii) upregulated the expression of HLA-DR and costimulatory molecules CD80 and CD86; iv) enhanced phagocytic potential of macrophages; v) enhanced CD8 T cell mediated target lysis; vi) the mechanism elucidated in curbing the survival of Mtb was through the involvement of nitric oxide and autophagy; vii) signaling pathway induced by IFG was through phosphorylation of STAT-3, Akt and activation of NF-κB.
For the optimum activation of T cells, two signals are required. First is the occupancy of T-cell receptor (TCR) by MHC-peptide complex. Second signal is also APCs driven in the form of costimulatory molecules like CD80 and CD86. In the absence of second signal, T cells undergo anergy, i.e. a state of unresponsiveness. Upregulation of HLA-DR, CD80 and CD86 by IFG indicate its potential role in the optimal activation of T cells. Further, it will help in counteracting Mtb-mediated downregulation of costimulatory molecules and thereby avoiding T cell anergy23. IFG predominantly promotes the generation of Th1 cells and CD8 T cells32,49,50,51. Both these cells are responsible for cell mediated immunity and play a cardinal role in protection against Mtb. CD8 T cells lyse the infected cells and release the intracellular Mtb. The IFN-γ secreted by Th1 cells activate macrophages and consequently they phagocytose and eliminate the bacterium. It was an exciting observation that IFG elicited macrophages showed enhanced phagocytosis and killing of Mtb. These results reveal a novel role of IFG against Mtb.
IFN-α/β interacts with IFNAR receptor and induces the activation of Jak-1 and Tyk-2. Jak-1 stimulates and phosphorylates STAT-338,39. Consequently, Akt-PI3K is activated through STAT-3 and translocates into the nucleus. Further, Akt-PI3K-STAT-3 signaling helps in delivering survival signal through the activation of NF-κB38,39. Like IFN-α/β, IFG [(IFN)-α2b] also exhibits a specific affinity for IFNAR and activates tyrosine kinase Jak-1 along with Tyk-238,39,52,53. It is also important to mention that signaling through IL-6 also follows the gp130/Jak/STAT pathway54. We investigated the signaling mechanism mediated by IFG in activating macrophages to restrict the intracellular survival of Mtb. We observed that Mtb infected macrophages when activated with IFG showed release of IL-6, but no noticeable change was seen in IL-1β, TGF-β, IL-10, TNF-α genes except IL-6 gene expression and subsequent phosphorylation of STAT-3 and Akt but not STAT-1. This indicates that IL-6 pathway is involved in IFG signaling. Phosphorylation of STAT-3 and Akt-PI3K delivers a survival signal to macrophages through upregulation of Bcl-xL and activation of NF-κB, thereby ensuring cell survival, activation and induction of autophagy. We speculate that STAT-3 binds to a promoter sequence of INOS gene, enhances the synthesis of iNOS and therefore production of NO that ultimately leads to the killing of Mtb [Fig. 7]. The data were validated by blocking the Akt-PI3K, iNOS and autophagy by their respective inhibitors to establish the role of autophagy in restricting the growth of Mtb by IFG stimulated macrophages. Blocking of Akt-PI3K, iNOS and autophagy, and substantial reduction in the secretion of NO and autophagy, as well as increase in the survival of Mtb signifies that autophagy is directly involved in the killing of Mtb. Further, the function of iNOS and autophagy are interlinked with each other. This provides the unambiguous evidence of a possible mechanism operating in constraining the Mtb growth. It has been reported that NO suppresses mTOR expression, which leads to autophagy55. Corroborating with this observation, we hypothesize that NO directly suppresses the expression of mTOR, as shown by the enhancement of NO release by mTOR inhibitor rapamycin (autophagy inducer), which led to the induction of autophagy in Mtb infected and IFG stimulated human macrophages. In essence, this study provides a novel immunomodulatory and therapeutic role of IFG against intracellular pathogen Mtb, which may have enough potential in future to treat TB patients.
Materials and Methods
Ethics statement
This study was permitted by the Institutional Biosafety and Ethical Committees of Institute of Microbial Technology, Chandigarh, India. All blood related work and experimental protocols were approved by the Institutional Biosafety and Ethical Committees of Institute of Microbial Technology, Chandigarh for project license IBSC8/IMT/10.7.12, and ethical guidelines for biomedical research on human subjects by Central Ethics Committee on Human Research (CECHR), ICMR-2000. Human blood was collected from healthy volunteers. The informed consent was obtained from all volunteers.
Reagents
Human Infergen was a kind gift from Dr. Eleanor Fish, The Department of Immunology, University of Toronto, Toronto, Canada.
THP-1 monocytes (ATCC TIB 202) were differentiated into macrophages by PMA (phorbol-12-myristate-13-acetate) (Calbiochem, San Diego, CA) for 16 h. The cells were washed 3x and rested for next 16 h in RPMI-1640 (GIBCO Invitrogen Corporation, Grand Island, NY) medium supplemented with FBS (10%), L-glutamine (2 mM), HEPES (10 mM), sodium bicarbonate (2.2 mg/ml), penicillin (0.7 mg/ml), streptomycin (1 mg/ml) at 37 °C/5% CO2. Human PBMCs were activated with PHA (2 μg/ml) (phytohemagglutinin PHA-M, Sigma-Aldrich, St. Louis, MO).
All the chemicals and reagents were purchased from Sigma Aldrich (St. Louis, MO) or otherwise mentioned. Free or fluorochrome conjugated antibodies (Abs): CD8-FITC, CD40 FITC, CD80-PE and PerCPefluor710, CD19-FITC, CD86-PE, HLA-DR-FITC and PEcy7, CD11b-APC, HLA-DR-PerCP 5.5, CD25-APC-Cy7 and the reagents used for cytokine ELISA were purchased from BD Pharmagin (San Diego, CA) and eBioscience (San Diego, CA). Fetal bovine serum (FBS) was purchased from Biological Industries (Kibbutz Beit Haemek 25115, Israel) and GIBCO (Grand Island, NY). L-pyruvate, L-glutamine, streptomycin and penicillin were from Serva (Heidelberg, Germany). Tissue culture grade plastic-ware was purchased from BD Biosciences (Bedford, MA). Abs used in Western blotting against pAkt (Thr308) [C31E5E], Akt (Pan) [C67E7] and PI3 kinase p85 (19H8) [4257] were purchased from Cell Signaling Technology (Danvers, MA). Abs to pSTAT-3 (pY705) [612357], STAT-3 [610190], pSTAT-1 (pY701) [612132] and STAT-1 [610115] were procured from BD Biosciences (Bedford, MA) and iNOS [Ab3523] from Abcam (Cambridge, UK). Inhibitors against STAT-3 [S3I-201], PI3K [Ly294002] were bought from Sigma Aldrich (St. Louis, MO), and Akt [Akt inhibitor IV, 124011], iNOS [NM, 475886] and NF-кB [481408] from Calbiochem (Billerica, MA). The GFP+ Mtb-H37Ra was a kind gift from Dr. Pawan Gupta, CSIR-Institute of Microbial Technology, Chandigarh, India.
Activation of THP-1 cells by IFG
THP-1 monocytes (3 × 105/well) were differentiated in 24 well plate with PMA (25 ng/ml) for 16 h. The cells were washed and then rested for next 16 h. Later, cells were stimulated with different concentrations of IFG (0–64 ng/ml) for 24 h.
T cell proliferation
PBMCs (1.5 × 105 cells/well) isolated from human blood by ficoll-diatrizoate (Histopaque-1077, Sigma-Aldrich, St. Louis, MO) density gradient centrifugation method were stimulated with PHA (2 μg/ml) and cultured with different concentrations of IFG (0–64 ng/ml) for 48 h in 96 well U-bottom plate. Later, 3H-thymidine (0.5 mCi/well) (Perkin Elmer, MA) was incorporated into the cultures and incubated for additional 16 h. Subsequently, the culture plates were harvested on glass-fiber filter mats by a Tomtec Harvester-96 (Hamden, CT). The radioactivity incorporated was measured using β-scintillation counter (Wallac-1450 Microbeta Trilux, Perkin Elmer, Waltham, MA) and expressed as counts per minute (cpm).
The activation of macrophages, B cells and T cells by IFG
PBMCs (2.5 × 105/well) were stimulated with IFG (64 ng/ml) in the presence or absence of PHA for 48 h. Later, cells were harvested and stained with fluorochrome conjugated Abs to detect the activation of macrophages (CD11b+/CD80+/CD86+/HLA-DR+), B cells (CD19+/CD80+/CD86+/HLA-DR+) and CTLs (CD8+/CD25+) and their isotype matched control Abs for 30 min/4 °C. Usual procedures of incubation and washings were followed at each step. Finally, cells were fixed in paraformaldehyde (1%). The flow cytometry data were acquired using FACS Calibur and FACS Aria-II and analyzed by DIVA and Flowjo softwares (BD Biosciences, San Jose and Williamson Way, Ashland, OR).
Mycobacterial uptake by IFG stimulated macrophages
THP-1 monocytes (3 × 105/well) were differentiated with PMA (25 ng/ml) for 16 h, followed by resting for additional 16 h in 24 well plate. Later, macrophages were stimulated with IFG (64 ng/ml) for 24 h in RPMI-FBS-10% at 37 °C/5% CO2. After 24 h, IFG stimulated cells were infected with Mtb strains H37Rv or H37Ra at MOI (1:5) for 4 h (3 h for M. smegmatis). This was followed by incubation with amikacin (2 μg/ml) for 1 h to kill extracellular bacteria. After 1 h, cells were lysed with saponin (0.1%) and lysate (100 μl) was plated on 7H11 agar plates. Bacterial colonies were enumerated on 21d. All subsequent experiments were performed with strain H37Rv (Mtb) or otherwise indicated.
Phagocytosis of GFP+ Mtb by IFG stimulated macrophages by confocal microscopy
Macrophages (3 × 105/well) were stimulated as mentioned above with IFG for 24 h. Later, cells were infected with GFP+ Mtb (H37Ra) [MOI:1:10] for 4 h. The amikacin (2 μ g/ml) treatment was given for 1 h to kill the extracellular bacteria, followed by extensive washing with cold PBS. Subsequently, cells were fixed with paraformaldehyde (1%) for 10 min. These cells were washed and placed in poly-L lysine pre-coated cover slips and imaged using Nikon A1 confocal laser microscope (Nikon, Tokyo, Japan). Z-stacks and extent of GFP+ Mtb internalization were monitored among experimental and control samples. The analysis of the intensity of surface plots was performed using Nikon NIS-AR 4.1 image analysis software (Nikon, Melville, NY). The confocal results were further validated by flow cytometry and analysis was done by FACS Suite software (BD Biosciences, San Jose).
Intracellular killing of Mtb by IFG stimulated macrophages
Macrophages were infected with Mtb (H37Rv or H37Ra) at MOI (1:10) for 4 h in 24 w plates, followed by extensive washings with PBS (1X) to remove extracellular bacteria. Later, cells were cultured in antibiotic free RPMI + FBS-10% along with IFG (0–64 ng/ml) for 24 h. Amikacin (2 μg/ml) was added to the cultures to kill the extracellular Mtb. After 24 h, cells were lysed with saponin (0.1%) and lysate (100 μl) was plated on 7H11 agar plate. Colonies were enumerated on 21d.
Cell culture and estimation of cytokines
THP-1 macrophages (2.5 × 105/w) and macrophages (2.5 × 105/w) isolated from the human PBMCs by adhering on tissue culture grade petri plates for 4–6 h were cultured with IFG (0–64 ng/ml) in 48 w plates for 48 h. The control cells were cultured in medium alone. The supernatants were collected after 24 h and IL-6 and IFN-γ were estimated. Briefly, 96 w plates were coated O.N/4 °C with Abs to human IL-6 (2 μg/ml) and IFN-γ (2 μg/ml) in phosphate buffer (pH 9.2, 0.01M). The unbound sites were blocked with BSA (1%) in PBS (1X) for 3 h/RT. Subsequently, respective recombinant cytokines as standard, and culture supernatants (50 μl volume) were added and incubated ON/4 °C. Later, biotinylated anti-human IL-6 (2 μg/ml) and IFN-γ (2 μg/ml) Abs diluted (1:1) in PBS-Tween buffer were added into the plates and incubated for 2 h/RT. Next, avidin-HRP (1:10,000) was added and incubated at 37 °C/45 min. The regular procedures of washing and incubation were followed at each step. The color was developed using H2O2-OPD substrate-chromogen. The reaction was stopped by H2SO4 (7%) after 20 min. The plates were read at 492 nm. The results expressed as pg/ml of secreted cytokines were calculated using serial log2 dilutions of standard curve drawn with rIL-6 and rIFN-γ.
Viability of IFG treated macrophages
Macrophages were stimulated with IFG (0–64 ng/ml) for 24 h at 37 °C in RPMI-FBS-10% (500 μl) in 48 w plates. The stimulated cells were harvested, washed and resuspended in PBS and stained with 5 μl/tube annexin V and 2.5 μl/tube of propidium iodide (50 mg/ml). The cells were incubated in the dark for 15 min at RT. Later, PBS (200 μl) was added to the cells and acquired immediately using BD FACS Calibur flow cytometer and analysis was done by DIVA software.
Real time PCR for the quantification of IL-6, ATG5, ATG7, BECLIN-1, INOS, IL-1β, IL-10, TGF-β and TNF-α
Total RNA was isolated using trizol reagent from IFG (64 ng/ml) stimulated macrophages for 6 h, according to the manufacturer’s instruction (Invitrogen, Carisbad, CA). Briefly, RNA was quantified with the help of NanoDrop spectrophotometer. A260/A280 ratio of all the samples was in the range of 1.90 to 2.00. DNA contamination from RNA samples was removed by amplification grade DNase. RNA samples (3 μg) were incubated with DNase (3U) for 15 min in the reaction buffer. Later, DNase activity was terminated by adding stop solution. Further, the samples were heated at 70 °C/10 min to inactivate DNase activity. Analysis was done by comparative Ct method, whereas Ct values were normalized against house-keeping control actin. The results are represented as relative expression (fold change). The fold change was calculated considering the value of uninfected/unstimulated controls as 1. RT-qPCR and data analysis were done by Step One Plus RT-qPCR system (Applied Biosystems, Chromos, Singapore). The RT-qPCR primer sequences are as follows:
β-ACTIN primer: β-ACTIN-F 5′-AGAGGGAAATCGTGCGTGAC-3′
β-ACTIN-R 5′-CAATAGTGATGACCTGGCCGT-3′
IL-6 gene primer: CIL-6-H R 5′-CAGGGGTGGTTATTGCATCT-3′
CIL-6-H R 5′-CAGGGGTGGTTATTGCATCT-3′
ATG5 gene primer: ATG5-Fwd 5′-AGAAGCTGTTTCGTCCTGTGG-3′
ATG5-Rev 5′-AGGTGTTTCCAACATTGGCTC-3′
ATG7 gene primer: ATG7-Fwd 5′-AATGTCTGCTGCTTGGAGCC -3′
ATG7-Rev 5′-ACCCCCTAGGCAATCTTCAAA-3′
BECLIN-1 gene primer: BECLIN1-Fwd 5′-ACCTCAGCCGAAGACTGAAG-3′
BECLIN1-Rev 5′-AACAGCGTTTGTAGTTCTGACA -3′
INOS gene primer: EiNOS-H F 5′-ACAAGCCTACCCCTCCAGA-3′
EiNOS-H R 5′-TCCCGTCAGTTGGTAGGTTC-3′
IL-1β gene primer: hIL-1β-RT F 5′-GGACAAGCTGAGGAAGATGC-3′
hIL-1β-RT R 5′-TCGTTATCCCATGTGTCGAA-3′
TGF-β gene primer: CTGF-B1H F 5′-CTGGAAACCCACAACGAAA-3′
CTGF-B1H R 5′-AACTTGAGCCTCAGCAGAC-3′
TNF-α gene primer: hTNFα-RT F 5′-CACCATGAGCACTGAAAGCA-3′
hTNFα-RT R 5′-CGAGAAGATGATCTGACTGCCT-3′
IL-10 gene primer: hIL10-RT F 5′-GAGAACAGCTGCACCCACT-3′
hIL10-RT R 5′-CCCAGGTAACCCTTAAAGTCC-3′
Target cell lysis by CD8 T cells
The target cells were prepared using PBMCs labelled with carboxyfluorescein succinimidyl ester (CFSE) (1 μM) for 8 min/37 °C in PBS (1X). The cells washed 3X with PBS-FCS 1% were γ-irradiated (3000R). Allogeneic PBMCs were isolated from second donor and T cells were purified and stimulated with IFG. Later, target and effector cells were cocultured (1:2 ratio). The percentage of CFSE-labeled PBMCs lysed by CD8 T cells were analyzed by flow cytometry. The formula was used to analyze specific lysis: ratio = (mean percentage of CFSEhi cells/mean percentage of CFSElo cells), and the percentage of specific lysis = [1 − (ratio for CFSEhi cells/ratio for CFSElo cells)] × 100.
Nitric Oxide (NO) production
Macrophages were infected with Mtb for 4 h and stimulated with IFG (64 ng/ml) for 24 h. Later on, supernatants were collected and NO was estimated by Griess method36. Briefly, supernatants (50 μl) were incubated with an equal volume of Griess reagent along with sodium nitrite as standard for 5–10 min/RT. Later, absorbance was measured at 550 nm. Further, the involvement of NO in IFG signaling was authenticated by treating the cells with iNOS inhibitor (NM: N-monomethyl-L arginine) (20 μM), Akt inhibitor IV (20 μM) and NF-кB inhibitor (50 μM) for 1 h and stimulated with IFG for 24 h.
Inhibition of iNOS expression by N-monomethyl-L arginine
Mtb infected Mφs were stimulated with IFG in the presence or absence of iNOS inhibitor (NM: N-monomethyl-L arginine) (20 μM) for 24 h. Later, cells were lysed and CFU plating was done to quantify the viable bacteria.
Cell cultures and Western blotting
The Mtb infected macrophages were incubated with IFG (64 ng/ml). Further, to prove specifity, the cultures were treated with specific inhibitors for STAT-3 [S3I-201] (50 μM), PI-3K [Ly294002] (20 μM), Akt (20 μM), iNOS [NM] (20 μM) and autophagy [3MA] (20 mM) for 1 h and stimulated with IFG (64 ng/ml) at different time-points [16 h for iNOS, 4 h for LC3B, 15–30 min for STAT-3 | Akt | STAT-1 | Bcl-xL | PI3K] in 12 w plate. Cells stimulated with LPS (2 μg/ml) were used as a positive control. These cells were then harvested, washed, and lysed in lysis buffer (Cytoplasmic protein extraction buffer, phosphatase and protease inhibitor cocktail). Later, protein was estimated in the lysate and SDS-PAGE was performed. After transfer to PVDF membranes and subsequent blocking, the membranes were immunoblotted using Abs to phosphorylated and non-phosphorylated form of STAT-3, Akt, STAT-1 and PI3K. Blots were also incubated with Abs against LC3B, Bcl-xL, iNOS and loading control actin. Blots were developed using a chemiluminescence kit (Amersham Pharmacia Biotech, Buckinghamshire, UK) (ECL; Pharmacia-Amersham, Freiburg, Germany). Scanning of the blots was done by ImageQuant LAS 4000 (GE Healthcare, Freiburg, Germany). Image analysis was performed with MultiGuage image and ImageJ analysis softwares.
Quantification of autophagy influx by confocal microscopy
The THP-1 macrophages were infected with GFP-H37Ra for 4 h and stimulated with IFG (64 ng/ml) and rapamycin [1 μM] for another 4 h. Mtb-Mφ-IFG were fixed on poly-L-lysine coated cover slips for 15 min. Later, cells were fixed with paraformaldehyde (2x) for 10 min followed by treatment with triton X100 (0.1%) for 3 min. The non-specific sites on cells were blocked with BSA (2%) for 2 h. This was followed by incubation with rabbit anti-human LC3 Ab for 2 h. Subsequently, cells were incubated with Alexa Fluor 633-anti-rabbit Ab for 1 h and followed by staining with DAPI dye. Regular washings were followed at each and every step. The cells were imaged through confocal microscopy. Rapamycin was used as a positive control. Number of puncta formation was counted manually from 4–5 different microscopic field. Percent of cells with LC3 puncta were presented in bar diagram.
Demonstration of NF-κB by EMSA
Macrophages were cultured with IFG for 30 min. Later, the cells were harvested and nuclear extract was prepared for EMSA. Briefly, an equal amount of nuclear extract (3 μg) from each sample was incubated for 20 min/37 °C in water bath with [32P] end labeled duplex oligonucleotides containing binding site for NF-κB. The DNA–protein complexes were resolved by electrophoresis on a native PAGE-gel (7%). After electrophoresis, the native gel was dried and exposed to screen at RT for 6–10 h and scanned by phospho-imager (Fujifilm, Tokyo, Japan).
The translocation of NF-κB by confocal microscopy
MφIFG were fixed on poly-L-lysine (0.1%) coated cover slips for 15 min. Later, cells were fixed with paraformaldehyde (2x) for 10 min followed by treatment with triton X100 (0.1%) for 3 min. The non-specific sites were blocked with BSA (2%) for 2 h. It was followed by incubation with mouse anti-human NF-κB p65 Ab for 2 h. Subsequently, cells were incubated with FITC-anti-mouse Ab for 1 h and followed by staining with Hoechst dye. Regular washings were followed at each step. The cells were imaged through Nikon A1 confocal laser microscope (Nikon, Tokyo, Japan) and analysis was performed using Nikon NIS-AR 4.1 image analysis software (Nikon, Melville, NY).
Statistics
The data analysis was done by Student’s ‘t’ test, non-parametric Mann-Whitney two tailed and repeated measure ANOVA with post Student-Newman-Keuls multiple comparison test using Graph Pad InStat 3 and Graph Pad Prism 6 softwares.
Additional Information
How to cite this article: Pahari, S. et al. Infergen Stimulated Macrophages Restrict Mycobacterium tuberculosis Growth by Autophagy and Release of Nitric Oxide. Sci. Rep. 6, 39492; doi: 10.1038/srep39492 (2016).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
World Health Organization. “Global tuberculosis report 2015.” 07/10/2015 (2015).
Lawn, S. D. & Zumla, A. I. Tuberculosis. Lancet 378, 57–72, doi: 10.1016/S0140-6736(10)62173-3 (2011).
Prabowo, S. A. et al. Targeting multidrug-resistant tuberculosis (MDR-TB) by therapeutic vaccines. Med Microbiol Immunol 202, 95–104, doi: 10.1007/s00430-012-0278-6 (2013).
Kaufmann, S. H. Tuberculosis vaccines: time to think about the next generation. Semin Immunol 25, 172–181, doi: 10.1016/j.smim.2013.04.006 (2013).
Isaacs, A. & Lindenmann, J. Virus interference. I. The interferon. Proc R Soc Lond B Biol Sci 147, 258–267 (1957).
Nagano, Y. & Kojima, Y. [Inhibition of vaccinia infection by a liquid factor in tissues infected by homologous virus]. C R Seances Soc Biol Fil 152, 1627–1629 (1958).
Feld, J. J. & Hoofnagle, J. H. Mechanism of action of interferon and ribavirin in treatment of hepatitis C. Nature 436, 967–972, doi: 10.1038/nature04082 (2005).
Wang, B. X. & Fish, E. N. The yin and yang of viruses and interferons. Trends Immunol 33, 190–197, doi: 10.1016/j.it.2012.01.004 (2012).
Chary, A. & Holodniy, M. Interferon combination therapy for HIV/hepatitis C virus coinfection. Immunotherapy 3, 1087–1102, doi: 10.2217/imt.11.105 (2011).
Clark, V. & Nelson, D. R. Novel interferons for treatment of hepatitis C virus. Clin Liver Dis 13, 351–363, doi: 10.1016/j.cld.2009.05.004 (2009).
Thitinan, S. & McConville, J. T. Interferon alpha delivery systems for the treatment of hepatitis C. Int J Pharm 369, 121–135, doi: 10.1016/j.ijpharm.2008.11.027 (2009).
Heathcote, J. Consensus interferon: a novel interferon for the treatment of hepatitis C. J Viral Hepat 5 Suppl 1, 13–18 (1998).
Trinchieri, G. Type I interferon: friend or foe? J Exp Med 207, 2053–2063, doi: 10.1084/jem.20101664 (2010).
Pandey, A. K. et al. NOD2, RIP2 and IRF5 play a critical role in the type I interferon response to Mycobacterium tuberculosis. PLoS Pathog 5, e1000500, doi: 10.1371/journal.ppat.1000500 (2009).
Manca, C. et al. Virulence of a Mycobacterium tuberculosis clinical isolate in mice is determined by failure to induce Th1 type immunity and is associated with induction of IFN-alpha/beta. Proc Natl Acad Sci USA 98, 5752–5757, doi: 10.1073/pnas.091096998 (2001).
Berry, M. P. et al. An interferon-inducible neutrophil-driven blood transcriptional signature in human tuberculosis. Nature 466, 973–977, doi: 10.1038/nature09247 (2010).
Koster, R. et al. Consensus-interferon and platelet-derived growth factor adversely regulate proliferation and migration of Kaposi’s sarcoma cells by control of c-myc expression. Am J Pathol 149, 1871–1885 (1996).
Gong, L., Devenish, R. J. & Prescott, M. Autophagy as a macrophage response to bacterial infection. IUBMB Life 64, 740–747, doi: 10.1002/iub.1070 (2012).
Klein, L., Munz, C. & Lunemann, J. D. Autophagy-mediated antigen processing in CD4(+) T cell tolerance and immunity. FEBS Lett 584, 1405–1410, doi: 10.1016/j.febslet.2010.01.008 (2010).
Lunemann, J. D. & Munz, C. Autophagy in CD4+ T-cell immunity and tolerance. Cell Death Differ 16, 79–86, doi: 10.1038/cdd.2008.113 (2009).
Castillo, E. F. et al. Autophagy protects against active tuberculosis by suppressing bacterial burden and inflammation. Proc Natl Acad Sci USA 109, E3168–3176, doi: 10.1073/pnas.1210500109 (2012).
Chan, E. D., Chan, J. & Schluger, N. W. What is the role of nitric oxide in murine and human host defense against tuberculosis? Current knowledge. Am J Respir Cell Mol Biol 25, 606–612, doi: 10.1165/ajrcmb.25.5.4487 (2001).
Khan, N., Gowthaman, U., Pahari, S. & Agrewala, J. N. Manipulation of costimulatory molecules by intracellular pathogens: veni, vidi, vici!! PLoS Pathog 8, e1002676, doi: 10.1371/journal.ppat.1002676 PPATHOGENS-D-11-02526 (2012).
Khan, N., Vidyarthi, A., Pahari, S. & Agrewala, J. N. Distinct Strategies Employed by Dendritic Cells and Macrophages in Restricting Mycobacterium tuberculosis Infection: Different Philosophies but Same Desire. Int Rev Immunol, doi: 10.3109/08830185.2015.1015718, [Epub ahead of print] (2015).
Fernando, M. R., Reyes, J. L., Iannuzzi, J., Leung, G. & McKay, D. M. The pro-inflammatory cytokine, interleukin-6, enhances the polarization of alternatively activated macrophages. PLoS One 9, e94188, doi: 10.1371/journal.pone.0094188 PONE-D-14-04332 (2014).
van der Poll, T. et al. Interleukin-6 gene-deficient mice show impaired defense against pneumococcal pneumonia. J Infect Dis 176, 439–444 (1997).
Godfrey, K. R. Chronopharmacology and its application to the development of theophylline treatment schedules for asthma. Eur J Clin Pharmacol 36, 103–109 (1989).
Nish, S. A. et al. T cell-intrinsic role of IL-6 signaling in primary and memory responses. Elife 3, e01949, doi: 10.7554/eLife.01949 (2014).
Diehl, S. A., Schmidlin, H., Nagasawa, M., Blom, B. & Spits, H. IL-6 triggers IL-21 production by human CD4+ T cells to drive STAT3-dependent plasma cell differentiation in B cells. Immunol Cell Biol 90, 802–811, doi: 10.1038/icb.2012.17 (2012).
Saunders, B. M., Frank, A. A., Orme, I. M. & Cooper, A. M. Interleukin-6 induces early gamma interferon production in the infected lung but is not required for generation of specific immunity to Mycobacterium tuberculosis infection. Infect Immun 68, 3322–3326 (2000).
Kropp, K. A. et al. Reversible inhibition of murine cytomegalovirus replication by gamma interferon (IFN-gamma) in primary macrophages involves a primed type I IFN-signaling subnetwork for full establishment of an immediate-early antiviral state. J Virol 85, 10286–10299, doi: 10.1128/JVI.00373-11 (2011).
Longhi, M. P. et al. Dendritic cells require a systemic type I interferon response to mature and induce CD4+ Th1 immunity with poly IC as adjuvant. J Exp Med 206, 1589–1602, doi: 10.1084/jem.20090247 (2009).
Martinez, J., Huang, X. & Yang, Y. Direct action of type I IFN on NK cells is required for their activation in response to vaccinia viral infection in vivo. J Immunol 180, 1592–1597, doi: 180/3/1592 (2008).
Platanias, L. C. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat Rev Immunol 5, 375–386, doi: 10.1038/nri1604 (2005).
Sakamoto, E. et al. Type I and type II interferons delay human neutrophil apoptosis via activation of STAT3 and up-regulation of cellular inhibitor of apoptosis 2. J Leukoc Biol 78, 301–309, doi: 10.1189/jlb.1104690 (2005).
Guevara, I. et al. Determination of nitrite/nitrate in human biological material by the simple Griess reaction. Clin Chim Acta 274, 177–188, doi: S0009898198000606 (1998).
Yuk, J. M. et al. Vitamin D3 induces autophagy in human monocytes/macrophages via cathelicidin. Cell Host Microbe 6, 231–243, doi: 10.1016/j.chom.2009.08.004 (2009).
Caraglia, M. et al. Alpha-interferon and its effects on signalling pathways within cells. Curr Protein Pept Sci 5, 475–485 (2004).
Pfeffer, L. M. et al. STAT3 as an adapter to couple phosphatidylinositol 3-kinase to the IFNAR1 chain of the type I interferon receptor. Science 276, 1418–1420 (1997).
Stetson, D. B. & Medzhitov, R. Type I interferons in host defense. Immunity 25, 373–381, doi: 10.1016/j.immuni.2006.08.007 (2006).
Jahrling, P. B. et al. Evaluation of immune globulin and recombinant interferon-alpha2b for treatment of experimental Ebola virus infections. J Infect Dis 179 Suppl 1, S224–234, doi: 10.1086/514310 (1999).
Julander, J. G., Ennis, J., Turner, J. & Morrey, J. D. Treatment of yellow fever virus with an adenovirus-vectored interferon, DEF201, in a hamster model. Antimicrob Agents Chemother 55, 2067–2073, doi: 10.1128/AAC.01635-10 (2011).
Brierley, M. M. & Fish, E. N. Review: IFN-alpha/beta receptor interactions to biologic outcomes: understanding the circuitry. J Interferon Cytokine Res 22, 835–845, doi: 10.1089/107999002760274845 (2002).
Adah, S. A., Bayly, S. F., Cramer, H., Silverman, R. H. & Torrence, P. F. Chemistry and biochemistry of 2’,5’-oligoadenylate-based antisense strategy. Curr Med Chem 8, 1189–1212 (2001).
Lande, R. et al. IFN-alpha beta released by Mycobacterium tuberculosis-infected human dendritic cells induces the expression of CXCL10: selective recruitment of NK and activated T cells. J Immunol 170, 1174–1182 (2003).
Fish, E. N., Banerjee, K., Levine, H. L. & Stebbing, N. Antiherpetic effects of a human alpha interferon analog, IFN-alpha Con1, in hamsters. Antimicrob Agents Chemother 30, 52–56 (1986).
Desmet, V. J., Knodell, R. G. et al. Formulation and application of a numerical scoring system for assessing histological activity in asymptomatic chronic active hepatitis [Hepatology 1981;1:431–435]. J Hepatol 38, 382–386, doi: S0168827803000059 (2003).
Alton, K., Stabinsky, Y., Richards, R. et al. Production, characterization and biological effects of recombinant DNA derived human IFN-α and IFN-β analogs. In: De Maeyer, E., Schellekens, H. eds The Biology of the Interferon System. Elsevier Science Publishers: Amsterdam. 119–128 (1983).
Cooper, A. M. Cell-mediated immune responses in tuberculosis. Annu Rev Immunol 27, 393–422, doi: 10.1146/annurev.immunol.021908.132703 (2009).
Everett, H. & McFadden, G. Apoptosis: an innate immune response to virus infection. Trends Microbiol 7, 160–165, doi: S0966-842X(99)01487-0 (1999).
Price, D. A., Klenerman, P., Booth, B. L., Phillips, R. E. & Sewell, A. K. Cytotoxic T lymphocytes, chemokines and antiviral immunity. Immunol Today 20, 212–216, doi: S0167-5699(99)01447-4 (1999).
Swanson, C. L. et al. Type I IFN enhances follicular B cell contribution to the T cell-independent antibody response. J Exp Med 207, 1485–1500, doi: 10.1084/jem.20092695 (2010).
Feld, J. et al. Mechanism of action of interferon and ribavirin in treatment of hepatitis C. Nature 436, 967–972, doi: 10.1038/nature04082 (2005).
Heinrich, P. C., Behrmann, I., Muller-Newen, G., Schaper, F. & Graeve, L. Interleukin-6-type cytokine signalling through the gp130/Jak/STAT pathway. Biochem J 334(Pt 2), 297–314 (1998).
He, H. et al. Nitric oxide induces apoptosis and autophagy; autophagy down-regulates NO synthesis in physalin A-treated A375-S2 human melanoma cells. Food Chem Toxicol 71, 128–135, doi: 10.1016/j.fct.2014.06.007 (2014).
Acknowledgements
We are thankful to Dr. Sathi Babu Chodisetti, Mr. Sudeep K. Maurya and Mr. R. Girish Nair, Ms Shikha Sharma for suggestions and helping in the preparation of the manuscript. Authors are thankful to Council of Scientific and Industrial Research for financial support. SP is a recipient of the fellowship of Council of Scientific and Industrial Research, NK and SN of Department of Biotechnology and MA of Department of Science and Technology, India.
Author information
Authors and Affiliations
Contributions
In this study the concept was generated and troubleshooting was solved by J.N.A. and S.P. The experiments were conducted by S.P., N.K., M.A., S.N. and J.K. The designing of experiments, data analysis and manuscript was written by S.P., J.N.A.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Pahari, S., Khan, N., Aqdas, M. et al. Infergen Stimulated Macrophages Restrict Mycobacterium tuberculosis Growth by Autophagy and Release of Nitric Oxide. Sci Rep 6, 39492 (2016). https://doi.org/10.1038/srep39492
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep39492
This article is cited by
-
NOS2/miR-493-5p Signaling Regulates in the LPS-Induced Inflammatory Response in the RAW264.7 Cells
Biochemical Genetics (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
