
Abstract
IL-18 is a member of the IL-1 family involved in innate immunity and inflammation. Deregulated levels of IL-18 are involved in the pathogenesis of multiple disorders including inflammatory and metabolic diseases, yet relatively little is known regarding its regulation. Liver X receptors or LXRs are key modulators of macrophage cholesterol homeostasis and immune responses. Here we show that LXR ligands negatively regulate LPS-induced mRNA and protein expression of IL-18 in bone marrow-derived macrophages. Consistent with this being an LXR-mediated process, inhibition is abolished in the presence of a specific LXR antagonist and in LXR-deficient macrophages. Additionally, IL-18 processing of its precursor inactive form to its bioactive state is inhibited by LXR through negative regulation of both pro-caspase 1 expression and activation. Finally, LXR ligands further modulate IL-18 levels by inducing the expression of IL-18BP, a potent endogenous inhibitor of IL-18. This regulation occurs via the transcription factor IRF8, thus identifying IL-18BP as a novel LXR and IRF8 target gene. In conclusion, LXR activation inhibits IL-18 production through regulation of its transcription and maturation into an active pro-inflammatory cytokine. This novel regulation of IL-18 by LXR could be applied to modulate the severity of IL-18 driven metabolic and inflammatory disorders.
Similar content being viewed by others


Introduction
Interleukin (IL)-18 and IL-1β are highly potent inflammatory cytokines that belong to the Interleukin-1 family of immunomodulators, and are implicated in a range of severe and diverse autoimmune and inflammatory diseases as well as metabolic and vascular disorders including rheumatoid arthritis, diabetes atherosclerosis1. IL-18 is primarily secreted by macrophages and dendritic cells2 and is constitutively expressed even in the absence of pro-inflammatory stimulation while IL-1β is absent from mononuclear cells and hematopoietic cells from healthy subjects3. One of the most prominent IL-18 actions is the induction of interferon gamma (IFNγ ) production by Th1 cells in the presence of IL-12 or IL-15 and thus is considered a pro-inflammatory cytokine. IL-18 also increases Fas Ligand (FasL) on natural killer (NK) cells thereby promoting FasL-mediated cytotoxicity.
IL-18 activity can be regulated at the (1) promoter/transcriptional level, (2) through post-translational processing (inflammasome-dependent cleavage) and (3) indirectly, through the sequestering role of the endogenous inhibitor IL-18 binding protein (IL18-BP)1. Despite its documented role on several pathologies, transcriptional regulation of the IL-18 gene remains largely unexplored. It has been previously shown that AP-1, PU.1, Interferon Regulatory Factor 8 (IRF8) as well as nuclear factor (NF)-κ B transcription factors play a critical role in the activation of the IL-18 promoter by LPS in macrophages4,5,6,7. Like IL-1β , IL-18 is produced as an inactive precursor (pro-IL-18) that is cleaved into a biologically active cytokine by the inflammasome protease component caspase 18,9. Inflammasomes are cytosolic multimeric complexes that are key to innate immune responses and consist of a sensor protein, an adaptor protein and the enzymatic component pro-caspase 1, which itself is subject to proteolytic activation. Inactive pro-caspase 1 is processed by the nucleotide-binding domain and leucine-rich repeat pyrin containing protein-3 (NLRP3) and apoptosis-associated speck-like protein (ASC) inflammasome complex formed upon LPS and adenosine-triphosphate (ATP) activation10,11. NLRP3 is triggered by many of the metabolic by-products generated in chronic metabolic diseases, including beta-amyloid plaques in Alzheimer’s disease, islet amyloid polypeptide involved in type 2 diabetes and cholesterol, hydroxyapatite and urea crystals involved in atherosclerosis, rheumatoid arthritis and gout, respectively12. The negative regulation of caspase 1 is thought to be crucial to a balanced inflammatory response, however this area of research is limited to a mere handful of studies13,14,15,16. A potent endogenous inhibitor of IL-18, IL-18 binding protein (IL-18BP) is also secreted to the ECM and directly interacts with IL-18 to inhibit its activity1. Most diseases implicating IL-18 actually result from an imbalance between IL-18 and IL-18BP circulating levels1. Thus, a finely tuned control of IL-18 and IL-18BP levels by a number of coordinated regulatory mechanisms is required. However, little is known on the gene regulation of IL-18BP levels17,18.
The nuclear receptors Liver X Receptors (LXRs) are lipid-activated transcription factors expressed in numerous immune cell types including macrophages whose activation modulates immune responses19. LXRs modulate gene transcription by heterodimerising with the Retinoid X Receptor (RXR) and binding to specific DNA sequences termed LXR response elements (LXREs) in the transcriptional regulatory regions of their target genes19. LXR transcriptional activity is induced by certain oxysterols and synthetic compounds including T090131720 or GW396521. Both LXR isoforms, LXRα and LXRβ , control macrophage cholesterol homeostasis and regulate macrophage inflammatory responses, phagocytosis and apoptosis22,23. LXR activation prevents macrophage cholesterol accumulation by inducing ABC transporter-mediated cholesterol efflux through the transcriptional regulation of ABCA1 and ABCG1 and limits cholesterol uptake. Furthermore, LXRs inhibit the transcription of pro-inflammatory genes (including IL-6, IL-1β and iNOS) lacking LXREs in their promoters or enhancers. They do so by antagonising the activity of transcription factors such as NF-κ B or AP1 without directly binding to DNA themselves, in a process termed transrepression23. IL-1β can be regulated by LXRs through this mechanism24 and in this way LXR activation inhibits inflammatory responses in macrophages and other immune cells including lymphocytes25. The postulated molecular mechanism underlying this regulation involves the SUMOylation of the receptor upon ligand binding, inhibiting LPS-induced corepressor clearance from the promoter of inflammatory genes26 although this mechanism has been recently challenged27. In addition to IL-1β , LXRs regulate multiple inflammatory, metabolic and vascular conditions where dysregulated IL-18 levels are observed1,25. However, whether LXR activation modulates IL-18 levels remains unknown.
In this study we demonstrate the novel negative regulation of IL-18 levels by LXR at multiple regulatory checkpoints: its expression, activation and bioavailability. We show for the first time that LXRs inhibit IL-18 gene expression and negatively regulate caspase 1 levels (and hence IL-18 processing and activation by the inflammasome). Moreover, through regulation of IRF8, LXR activation enhances the expression of IL-18BP thus also identifying IL-18BP as a novel IRF8 and LXR target gene. Collectively, LXRs exert multiple actions on IL-18 and IL-1β regulation.
Results
IL-18BP is a novel LXR target gene
IL-18 binding protein (IL-18BP) is a potent IL-18 endogenous inhibitor secreted by macrophages that directly neutralises IL-18 with a high level of affinity (Kd = 400 pM) at a 1:1 stœchiometry1. We assessed the effect of ligand-activated LXRs on IL-18BP gene expression in BMDM. BMDM offer advantage over other murine in vitro macrophage models as activation experiments can be performed on a homogenous population of quiescent cells28. Treatment with GW3965 ligand increased IL-18BP mRNA expression by almost 6-fold compared to control mouse macrophages (Fig. 1a). This effect was further enhanced by the RXR agonist LG268 in wild type (WT), but not in LXR-deficient, macrophages (Fig. 1b). Consistently, LXR ligand enhanced secretion of IL-18BP by macrophages (data not shown) and increased intracellular IL-18BP protein levels in these cells in the presence of Brefeldin A, an inhibitor of Golgi apparatus secretion (Fig. 1c). Thus, LXR activation enhances IL-18BP expression at the mRNA and protein level in primary macrophages in an LXR-dependent manner.

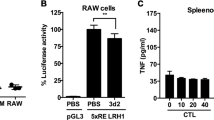
(a) BMDM were treated with vehicle (DMSO) or GW3965 (GW) for 24 hours and IL-18BP mRNA content was analyzed by RT-qPCR. Values indicate expression normalized to cyclophilin and are presented relative to the expression in vehicle-treated cells. Values represent the mean of 4 independent experiments ± SEM. t-test: ***p ≤ 0.001. (b) BMDM from WT or LXRα β deficient mice (LXRα β −/−) were treated as in A with or without the RXR activator LG268 (LG). IL-18BP mRNA content was analyzed as in A. Values represent the mean of 3 independent experiments ± SEM. *p ≤ 0.05, **p ≤ 0.01. (c) BMDM were treated with vehicle (DMSO) or GW3965 (GW) for 24 hours supplemented with Brefeldin A for the last 6 hours. Protein content was analyzed by immunoblotting and Hsp90 levels were assayed as loading control. (d) RAW264.7 cells were transfected with hLXRα or pcDNA3.1 (− ) along with the indicated IL-18BP luciferase reporters or pGL3 empty vector. Cells were treated as described in the Methods section. For each reporter, luciferase and β -galactosidase activities were measured and the ratio was compared to the vehicle-treated condition in the absence of LXRα (− ), which was set as 1. Data are mean value ± SD (n = 3) of one representative experiment. t-test: **p ≤ 0.01, ***p ≤ 0.001. (e) Upper panel, location of a putative LXRE in the IL-18BP locus. Arrows indicate the position of primers used for ChIP assays. Lower panel, BMDM cells were incubated with or without GW3965 at 1 μ mol/L for 2 hours. LXR occupancy was assessed by ChIP assays. Primers shown in upper panel were used to amplify an unrelated site at − 4.7 kb that served as a negative control, the − 1.1 kb putative LXRE, − 0.5 kb and TSS as GW3965 responsive sites. SREBP1c primers were used as a positive control for LXR binding. Shown is a representative experiment of three.
Nuclear receptors LXRα and LXRβ are ligand-activated transcription factors that often bind directly to regulatory regions in their target genes to activate their transcription. To further understand the LXR-mediated molecular mechanism underlying the regulation of IL-18BP gene expression, we first assessed the effect of GW3965 on IL-18BP promoter activity. RAW264.7 macrophage-like cells were transfected with an LXRα expression vector together with luciferase reporters driven by various fragments of the IL-18BP promoter and were subsequently activated with the LXR ligand GW3965 (Fig. 1d). IL-18BP promoter activity was specifically enhanced by the LXR ligand primarily in LXRα -overexpressing cells. The activity of the IL-18BP promoter-driven reporter with only 116 bp upstream of the transcription start site (TSS) however, was enhanced by LXRα even in the absence of ligand and was further increased in the presence of GW3965. This suggests the shorter IL-18BP promoter fragment is more sensitive to LXRα overexpression. We then assessed whether LXRs directly bind this LXR and GW3965-responsive region in the IL-18BP promoter by chromatin immunoprecipitation (ChIP) assays, compared to the LXRE described for SREBP1c29. A region at − 4.7kb in the IL-18BP gene was used as a negative control. Unexpectedly, LXRs did not bind either the IL-18BP proximal promoter region (TSS and − 0.5kb), or the − 1.1kb region bearing a strong putative LXR Response Element (Fig. 1e). By contrast, LXR occupied the SREBP1c promoter in vehicle- and ligand-activated cells as expected. Altogether, these results demonstrate that LXRα increases IL-18BP expression by activating the proximal IL-18BP promoter without directly binding this region, suggesting an additional transcription factor is involved in the regulation of IL-18BP by LXR.
LXR-mediated regulation of IL-18BP expression is dependent on IRF8
We have previously shown that the hematopoietic transcription factor IRF8 mediates the LXR ligand induced expression of arginase 1 in macrophages, in an LXR binding independent manner30, a regulation that may contribute to the anti-inflammatory properties of LXRs31. LXR ligand induction of IL-18BP mRNA levels is abolished in cells with knocked-down levels of this factor or lacking IRF8, demonstrating this transcription factor mediates LXR activation of IL-18BP expression (Fig. 2a,b). Interestingly, IL-18BP up-regulation by LXR persists even in a pro-inflammatory environment (Fig. S1a). The decrease in IL-18BP upon LPS treatment is associated with reduced levels of IRF8 expression in these cells, thus emphasizing the role of IRF8 on the regulation of IL-18BP (Fig. S1b). However, when macrophages were primed with low levels of IFNγ , expression of IL-18BP and IRF8 was not altered while LXR ligand induced their transcription. These data suggest that different inflammatory pathways affect basal and LXR ligand-activated IL-18BP expression in a different manner, which could have consequences when modulating IL-18 active levels.

(a) BMDM were transfected with control (Ctrl) or IRF8 siRNAs for 24 hours and treated vehicle (DMSO) or GW3965 (1 μ mol/L) for 24 hours. IL-18BP mRNA content was analyzed by RT-qPCR. Values indicate expression normalized to cyclophilin and are presented relative to the expression in siRNA-control vehicle-treated cells, which was set as 1. A representative experiment is shown (mean ± SD) t-test: ***p ≤ 0.001. (b) BMDM from WT or IRF8−/− mice were cultured with T1317 (T) (1 μ mol/L) for 24 hours. IL-18BP mRNA content was analyzed by RT-qPCR. Values indicate expression normalized to cyclophilin and are presented relative to the expression in siRNA-control vehicle-treated cells, which is set as 1. A representative experiment is shown (mean ± SD) t-test: **p ≤ 0.01. (c) BMDM were incubated as in Fig. 1E. IRF8 occupancy was determined by ChIP assays. Primers shown in Fig. 1E were used to amplify the − 0.5 kb and TSS putative IRF8 binding sites. Primers amplifying Cystatin C (CystC) and β -actin (β -act) were used as positive and negative controls, respectively. Background occupancy using IgG in the absence or presence of ligand is depicted by light and dark grey bars respectively. Shown is a representative experiment. (d) RAW-LXRα cells were treated with vehicle (DMSO) or GW3965 (GW) (1 μ mol/L) for 24 h. IRF8 was immunoprecipitated and tyrosine phosphorylation of IRF8 was analysed by immunoblotting. Protein content in the cell lysate prior to immunoprecipitation (Input) was analysed. A representative experiment is shown.
In silico analysis of the IL-18BP promoter sequence identified two putative Interferon Sensitive Response Elements (ISREs) located at the TSS and − 0.5kb upstream of the TSS. As previously shown, IRF8 binding to Cystatin C, an ISRE-containing gene32, was observed in the absence of LXR ligand and was further enhanced upon LXR activation (Fig. 2c), a likely consequence of the increase in IRF8 expression by LXR30 (and Fig. S1). While IRF8 was not detected in the IL-18BP promoter at both putative sites tested in vehicle-treated cells, IRF8 occupancy strongly increased upon LXR activation (Fig. 2c). This demonstrates that LXR activation enhances IRF8 binding to the IL-18BP promoter, ultimately regulating IL-18BP levels. We previously showed that LXR directly regulates IRF8 expression and promotes IRF8 heterodimerisation with the transcription factor PU.130. Dimerisation of IRF8/PU.1 and binding to their target genes is enhanced by phosphorylation of IRF8 at Tyr211 in LPS-activated macrophages2. Interestingly, IRF8 tyrosine phosphorylation was enhanced by the GW3965 ligand (Fig. 2d), suggesting that regulation of IRF8 tyrosine phosphorylation may also be an important component in the overall regulation of IRF8 activity by LXR.
LXRs inhibit LPS-induced IL-18 levels
LPS stimulation enhances the expression of IL-1 family members such as IL-1β and IL-18 through the activation of a PARP1-ERK-NF-κ B-dependent pathway in mouse macrophages33. As LXRs antagonise NF-κ B-mediated activation of pro-inflammatory genes in these cells24,26, we hypothesized these receptors also regulate IL-18 expression. IL-18 mRNA levels increased in the presence of LPS as described34, confirming a robust inflammatory response in BMDM. In LPS-activated macrophages, IL-18 mRNA levels were significantly inhibited upon LXR ligand activation by GW3965 compared to cells cultured in vehicle-supplemented medium (Fig. 3a). This was abolished in the presence of the LXR antagonist GSK144023335 or in macrophages from LXRα β −/− mice (Fig. 3b) demonstrating this inhibition is exerted in an LXR-dependent manner. LXR activation also diminished IL-18 gene expression in IFNγ -stimulated macrophages (Fig. S2a,b), further corroborating the inhibitory effect of LXR on pro-inflammatory cytokine-stimulated IL-18 expression. IL-18 is a secreted cytokine and its maturation and secretion is in part dependent on the activation of the NLRP3 inflammasome. LXR agonists also reduced the intracellular LPS-induced expression of the 24 kDa precursor form (pro-IL-18) and cleaved (active) IL-18 protein expression in the presence of ATP, a known NLRP3 inflammasome activator36 (Fig. 3c and S3a). This effect was also dependent on LXR activity as it was abolished in the presence of the LXR antagonist (Fig. 3d). Consistently, LXR ligands significantly decreased LPS and ATP-induced IL-18 levels in macrophage supernatants in a similar manner (Fig. 3e and S4b). Altogether, these results demonstrate that LXR inhibits mRNA and protein expression of pro-IL-18 and secreted levels of bioactive IL-18 in primary macrophages.

(a) BMDM were treated with vehicle (DMSO), GW3965 (1 μ mol/L) or LXR antagonist (LXR ant, 1 μ mol/L) for 24 hours with or without LPS (100 ng/ml) for the last 6 hours. IL-18 mRNA levels were analyzed by RT-qPCR. Values indicate expression normalized to cyclophilin and are presented relative to the expression in LPS-treated cells, set as 1. Values represent the mean ± SEM (3–4 different BMDM preparations). t-test: ***p ≤ 0.001, ns p > 0.05. (b) IL-18 mRNA expression in BMDM from WT and LXRα β −/− mice were analyzed by RT-qPCR. Shown is a representative experiment performed in triplicate (mean ± SD) t-test: *p ≤ 0.05, ns p > 0.05. (c) BMDM were treated as in A and then activated with ATP for the last 2 hours. Pro-IL-18 expression was analyzed by immunoblotting and Hsp90 levels were assayed as loading control. Shown is a representative experiment of three. (d) BMDM were treated as in A. Intracellular expression of IL-18 is shown as analyzed by immunoblotting. Samples shown were analysed on non consecutive lanes of the same blot. (e) BMDM were treated as indicated. Secreted IL-18 levels in culture supernatants were quantified by ELISA. Values represent mean ± SEM (n = 3). t-test: *p ≤ 0.05.
LXRs regulate IL-18 maturation
Processing the inactive pro-IL-18 precursor into a cleaved active IL-18 form in response to pro-inflammatory signals is exerted by a number of processing proteases. One of the best characterized is caspase 1, which cleaves and thus promotes the release of IL-18 from activated macrophages8,9. We hypothesized that irrespective of the transcriptional repression of the 24 kDa IL-18 precursor, which consistently occurs even when caspase 1 is inhibited (Fig. S4a), LXR activation affects IL-18 maturation by modulating caspase 1. Treatment with the LXR specific agonist GW3965 significantly inhibited caspase 1 gene expression in LPS-activated macrophages in an LXR-dependent manner, as the repressive effect was impaired in the presence of an LXR antagonist or in macrophages lacking LXRα β (Fig. 4a,b). Caspase 1 activation itself is regulated by the canonical inflammasome constituted by NLRP3/ASC10,11. In response to LXR ligand activation, intracellular expression of both the 45 kDa pro-form and the 20 kDa cleaved form of caspase 1 was reduced in the presence of the inflammasome activator ATP, suggesting that part of the reduction in cleaved IL-18 levels is likely due to diminished levels of caspase 1 (Fig. 4c and S3b,c). Interestingly, the regulation of the pro-caspase 1 form by the GW ligand was blocked by the LXR antagonist (Fig. S4d), demonstrating LXR-dependent effects by GW in these cells. Finally, in contrast to the regulation of IL-18 and caspase 1, NLRP3 expression was unaffected by LXR ligand activation (Fig. 4c–e), indicating the LXR inhibition of IL-18 is independent of a direct effect on the NLRP3 component of the inflammasome. Overall, our results demonstrate that LXR activation decreases IL-18 maturation in part by downregulating caspase 1 expression and activation.

BMDM were treated with vehicle (DMSO), GW3965, LXR antagonist (LXR ant) or LPS as indicated in Fig. 3A. (a) Caspase 1 mRNA expression was analyzed by RT-qPCR. Values indicate expression normalized to cyclophilin and are presented relative to the expression in LPS-treated cells. Values represent the mean ± SEM (n = 3–4). t-test: *p > 0.05, ns p > 0.05. (b) Caspase 1 mRNA levels in BMDM from WT and LXRα β −/− mice were analyzed by RT-qPCR. Shown is a representative experiment performed in triplicate (mean ± SD) t-test: *p ≤ 0.05, ns p > 0.05. (c) BMDM were treated with or without GW3965 and LPS for the final 6 hours with addition of ATP for the last 2 hours. Pro-caspase 1, cleaved caspase 1 and NLRP3 protein content were analyzed by immunoblotting and Hsp90 levels were used as loading control. (d) NLRP3 mRNA expression in LXR-ligand treated BMDM was analysed as in B. Shown is a representative experiment of 3 (mean ± SD). (e) NLRP3 protein levels were analyzed by immunoblotting. For (c,e), representative experiments of 2–3 independent assays are shown.
LXRs inhibit LPS-induced IL-1β levels in bone marrow-derived macrophages
Another IL-1 family member whose expression is enhanced by LPS stimulation in an NF-κ B-mediated fashion is IL-1β 33. LXR ligands have been shown to antagonize the activation of IL-1β in thioglycolate-ellicited peritoneal macrophages24,26,37. We next examined whether, in addition to IL-18, LXR activation modulates IL-1β expression in BMDMs, which are morphologically and phenotypically different from peritoneal macrophages and present a different cytokine expression profile38. LPS-stimulated IL-1β mRNA and secreted protein levels were inhibited upon LXR ligand activation by GW3965 in BMDMs (Fig. 5a,b) demonstrating that LXR regulated pathways involved in the production and maturation of IL-18, are also affecting IL-1β levels. In addition, LXRα β -deficient macrophages secrete enhanced levels of both IL-18 and IL-1β (Fig. S5a,b) further corroborating the role of LXR in the regulation of these inflammasome-activated cytokines.

(a) BMDM were treated with vehicle (DMSO), GW3965 (1 μ mol/L) or LXR antagonist (LXR ant) for 24 hours with or without LPS (100 ng/ml) for the last 6 hours. IL-1β mRNA levels were analyzed by RT-qPCR. Values indicate expression normalized to cyclophilin and are presented relative to expression in LPS-treated cells, set as 1. Values represent the mean ± SEM (n = 3–4). t-test: ***p < 0.001, ns p > 0.05. (b) Secreted IL-1β levels in cell supernatants were quantified by ELISA. Values represent the mean ± SEM (n = 3) **p < 0.01.
Overall, this study shows for the first time that LXR activation inhibits IL-18 production in macrophages through regulation of its transcription and processing into an active pro-inflammatory cytokine, as well as by inducing the expression of its potent endogenous inhibitor IL-18BP through the regulation of IRF8. This could have important consequences in the modulation of various inflammatory, metabolic, and vascular disorders by LXR.
Discussion
IL-18 is a potent pro-inflammatory molecule implicated in a number of inflammatory and autoimmune diseases including rheumatoid arthritis, multiple sclerosis and psoriasis as well as inflammatory and metabolic diseases including atherosclerosis1,39. IL-18 is a member of the IL-1 family of central mediators of innate immunity and inflammation and thus its tight regulation via receptor antagonists, decoy receptors and signaling inhibitors is needed to ensure the right balance between amplification of innate immunity and uncontrolled inflammation1. However, there is relatively little insight into its negative regulation. LXRs modulate the severity of conditions where dysregulated IL-18 levels are observed25, therefore we hypothesized that LXRs may affect these partly by regulating IL-18. In this report we describe for the first time IL-18 regulation at multiple points by LXRs, including its transcription, activation and bioavailability through regulation of the endogenous IL-18 inhibitor IL-18BP.
Our studies have primarily focused on the regulation of IL-18 in mouse bone marrow derived macrophages, which have been previously used to investigate LXR mode of action27,28,29. The majority of work studying the role of LXRs has been performed in mouse cells and despite some reported species-specific differences, important similarities on LXR actions between human and mouse cells exist40. Interestingly, experiments performed in human monocyte-derived macrophages suggest the actions reported here are relevant to a human setting (Fig S6).
LXR agonists robustly inhibited macrophage IL-18 mRNA levels and promoted IL-18BP levels and these effects were shown to be specific for LXR through the use of a combination of LXR-deficient cells and specific antagonists. Furthermore, LXR activation elicited a significant reduction in the LPS-stimulated pro-protein and in the cleaved and secreted bioactive levels of IL-18. In addition, IL-18BP is regulated by the GW ligand in human macrophage cells in an LXR-dependent manner (Fig. S6a) and the expression of the IRF8 mediator of this regulation is also induced in response to LXR ligands as previously shown30 (Fig. S6b). In agreement with our findings in mouse macrophages and LXR being a regulator of IL-18 and IL-1β , LPS-activated levels of these cytokines are exacerbated in human macrophages in the absence of the LXR receptors (Fig. S6c,d). This confirms previous studies demonstrating that LXR inhibits the expression of IL-1β as well as other cytokines and chemokines in LPS-stimulated human lung macrophages and the THP1 human monocytic cell line41. This work led to the suggestion that LXR ligands could be used for the treatment of lung diseases involving chronic lung inflammation mediated by macrophages. Indeed, LXR activation by GW3965 has shown anti-inflammatory effects (reduction of CXCl10 and CCL5 levels) in lung macrophages from chronic obstructive pulmonary disease patients42. In addition to responses to pro-inflammatory stimuli, LXRs have been reported to affect human lymphocyte activation and proliferation, dendritic cell migration, macrophage bacterial and viral infection and their phagocytic capacity to efficiently clear apoptotic cells (reviewed in40). Further supporting a role of LXRs in human immunity, a number of studies have linked LXR gene variants with immune disorders including susceptibility to tuberculosis, inflammatory bowel disease, systemic lupus erythematosus and rheumatoid arthritis40. However, the functional effects of some of the variants reported remain to be elucidated.
Previous investigations into the negative regulation of IL-18 are limited to observations of indirect mechanisms through caspase 1 modulation43 or inhibition by IL-18BP1. Our study demonstrates the direct negative regulation of IL-18 gene and protein expression. This work also extends previous findings demonstrating that LXR-mediated regulation of IL-1β is not limited to activated peritoneal macrophages.
Our data further reveal a modulatory role for LXR regulation on the inflammasome through the modulation of caspase 18,9. LXR activation significantly and specifically reduced mRNA and protein expression of the precursor form of caspase 1. Inflammasome regulation must be tightly controlled. However, previous investigations on the negative regulation of this complex have mainly focused on inhibitors of the sensor protein e.g. NLRP3, with limited reports on the transcription of the individual components (reviewed in43). Studies documenting negative regulation of caspase 1 are also scarce13,14,15,16. Recently, LXR was reported to induce caspase 1-dependent cell death in human colon cancer cell lines providing a mechanistic basis underlying some of the anticancer actions of LXR44. The mechanism involved however, is fundamentally different from the one we are proposing now for LPS-activated macrophages. According to this study, colon cancer cell death by LXR ligands implicated the non-transcriptional regulation of caspase 1 activation through the direct interaction of LXRβ with the membrane channel pannexin-1, leading to an induced release of ATP and subsequent pyroptosis or caspase 1-induced cell death. Of note, this study involves LXR activation with high concentrations (20 μ mol/L) of the less specific LXR agonist T090131720,45. In addition, this membrane channel has been shown to be required for ATP release in cells undergoing apoptosis but not for inflammasome activation per se46. Other differences between this report and our study are likely due to cell type, cell proliferating status, agonist type and/or the agonist concentration employed. We have used the most specific synthetic LXR agonist, GW396521,25, at a lower concentration (1 μ mol/L), in order to avoid off-target effects. It is worth noting that IL-18 processing is not exclusive of caspase 1 since other caspases produce a bioactive IL-1847. Following recognition of Gram-negative bacteria, caspase 11 activation (caspase-4 in humans) results in non-canonical inflammasome activation and the subsequent release of IL-1848. Additionally, processing of IL-18 can also be exerted upon FasL stimulation in the absence of caspase 149. Whether caspase 11 or FasL are also targeted by LXRs remains to be investigated.
The transcription factors that play a critical role in the activation of the IL-18 by LPS in macrophages include AP-1 and the hematopoietic transcription factors PU.1 and IRF84,5. IRF8 is a critical factor in lineage determination and the development of myeloid cells from common progenitor cells50,51,52. Indeed, IRF8 deficiency in mice is characterised by the massive accumulation of immature myeloid cells promoting the development of a chronic myelogenous leukemia-like phenotype51 and in humans, loss of function of IRF8 leads to monocytic and dendritic cell immunodeficiency53. Additionally, IRF8 is an important mediator of transcriptional responses to LPS and IFNβ 54, reflected by the high susceptibility to intracellular pathogens shown in IRF8-deficient mice51.
We previously showed that LXRα affects gene expression through IRF8 by directly inducing IRF8 levels and promoting IRF8 dimerization with PU.130, a mechanism that may contribute to the anti-inflammatory properties of LXRs31. We now demonstrate that LXR activation enhances IL-18BP expression in an IRF8-dependent manner, thus further implicating this hematopoietic factor as an important modulator of LXR actions beyond its role as a positive mediator of LPS and IFNβ responses. As we showed before for arginase 130, LXR agonists induced the binding of IRF8 to newly identified binding sites on the IL-18BP promoter that are otherwise unoccupied by these factors in untreated conditions. This suggests that LXR activation may expand the IRF8 cistrome, which may have important implications on disease progression when exposing macrophages or possibly other immune cell types to LXR ligands. Indeed, IRF8 was recently shown to bind distinct sets of binding sites, in resting macrophages and in response to LPS stimulation54. Whether a similar genome wide activation or redistribution of IRF8 binding sites occurs in response to LXR activation remains to be investigated. Specifically for macrophage IL-18 signalling, a differential activation of binding sites by LXR could explain how the same transcription factor, IRF8, is implicated both in the induction of IL-18 gene expression in response to LPS4,5 as well as in the expression of its endogenous inhibitor IL-18BP. This IRF8 upregulation of the potent IL-18 endogenous inhibitor elegantly ties in with the LXR reduction of IL-18 expression and activation demonstrated, illustrating an unprecedented negative regulatory role for LXR on this pro-inflammatory cytokine (Fig. 6).

Schematic representation of multiple negative regulatory points of LXR activation on IL-18 expression, maturation and bioavailability.
Transcription of inflammasome genes and subsequent IL-18 maturation is primarily regulated by Toll-like receptor (TLR)-dependent pathways55. LXRs have been shown to negatively regulate TLR signaling (namely, TLRs 2, 4 and 9) in mouse macrophages23 and they likely reduce IL-18 mRNA expression by interfering with these pathways. LXR activation has been shown to dampen LPS -driven inflammation24 through a mechanism termed ‘transrepression’ that does not involve the direct binding of LXR to promoter DNA. This involves the SUMOylation of LXR in response to its agonist, which leads to the stabilisation of NCoR transcriptional corepressor complexes on NF-κ B-bound promoters of inflammatory genes, thus actively inhibiting NF-κ B target gene transcription26. An alternative mechanism was recently proposed that rather involves changes in cellular lipid metabolism and local membrane composition and organisation27. This can occur in the absence of LXR SUMOylation and requires the expression of the cholesterol transporter ABCA1, which is a well characterised positively regulated LXR target gene. It was also shown that ABCA1 represses TLR-induced NFkB and MAPK activation by altering the cholesterol content of membrane domains. Further investigations will be required to identify the precise mechanisms responsible for the repression of the IL-18 and Caspase 1 precursors by LXR described in this study.
In summary, this report offers multiple fundamental new insights into the anti-inflammatory mechanisms of LXRs through the negative regulation of IL-18 at the level of transcription, protein activation and its bioavailability after secretion. This raises the therapeutic potential of LXR to treat the multiple diseases in which dysregulated IL-18 levels contribute to.
Methods
Cell culture and animal models
RAW264.7 or RAW-LXRα cells overexpressing LXRα were generated as described in56. Bone Marrow-derived Macrophages (BMDM) from C57BL/6 mice were prepared as in30 or57 using L929 Conditioned Medium (or LCM) as a source of M-CSF for the differentiation of the macrophages. LXRα β −/− mice58 were kindly provided by Dr. David Mangelsdorf (UT Southwestern). Expression of macrophage markers as assessed by flow cytometry analysis was similar in wild-type or LXR-deficient macrophage cultures (data not shown). After 7 days of differentiation, LCM-containing medium was removed, cells were washed with PBS and were then allowed to rest a minimum of 16h in DMEM containing low-endotoxin (≤ 10 EU/mL) FBS and 20 μ g/mL gentamycin without any LCM (activation medium) before being treated with DMSO, 1 μ mol/L T1317 (Sigma) or 1 μ mol/L GW3965 (Tocris) with or without 1–5 μ mol/L LXR antagonist (GSK1440233)35. All animal experiments were done in accordance to guidelines and regulations approved by the United Kingdom Home Office. For human monocyte-derived macrophage preparation, human peripheral blood mononuclear cells were isolated from blood of healthy donors by Ficoll density gradient centrifugation. CD14+ monocytes were isolated using the EasySep CD14+ Positive Selection Kit (Stemcell Technologies) following the manufacturers protocol or PBMCs were cultured directly in RPMI 1640 supplemented with 10% human serum and hMCSF (10–30 ng/mL, R&D Systems) for 7 days at 37 °C, 5% CO2 in order to obtain differentiated macrophages as in59. Cells were subsequently transfected with siRNA against LXRα β or non-specific scrambled RNA using Dharmafect 4 for 24 hours and activated as indicated. Human cell studies were performed in accordance with approved guidelines by the Ethics Committee of the University College London Hospitals National Health Service Trust. Healthy donors were recruited following their informed consent.
RNA analysis
BMDM were treated with vehicle, T1317 (1 μ mol/L) or GW3965 (1 μ mol/L) overnight with or without GSK antagonist (1 μ mol/L), followed by LPS activation (EColi 0127:B8 or Salmonella enterica serotype typhimurium, 100 ng/mL) for 3–6 hours. RNA was extracted and reverse transcription was performed as described30 using specific primers (Supplementary Table 1). Gene expression was determined as previously30. Each PCR determination was performed in duplicate and each experiment was conducted at least twice as indicated. For human macrophages, cells were activated with LPS (100 ng/ml, EColi 026:B6, Sigma) 24 hours after they were fully differentiated.
Promoter analysis
Luciferase assays were performed as described30. Luciferase reporters containing various fragments of the IL-18BP promoter were a kind gift from Dr. Bysani Chandrasekrar (U. Tulane).
Protein analysis
BMDMs were treated with vehicle or GW3965 (1 μ mol/L) for 24 h in activation medium, followed by Brefeldin A (3.5 μ mol/L) (for IL-18BP) and/or LPS for 6–24 h. Cells were lysed in RIPA buffer with protease inhibitors and protein content was analyzed by Western-blotting. Proteins in supernatants were precipitated using 20% tricholoroacetic acid and incubated overnight at − 20 °C. Antibodies against IL-18BPc (R&D systems, AF129), Hsp90 (Santa Cruz, sc-59577), IL-18 (Biovision, 5180), NLRP3 (Adipogen, MAB7578), Caspase 1 (Millipore 06–503, or Santa Cruz sc-514) and Tubulin (Sigma, T6074) were used. For ELISAs and immunoblotting of supernatants, BMDMs were cultured with vehicle or GW3965 for 6 h and medium was replaced with OptiMEM with LPS (500 ng/mL) and vehicle or GW3965 (1 μ mol/L) for a further 24 hours. ATP was added to a final concentration of 5 mmol/L for the last 2 h. IL-18 and IL-1β levels were analyzed by ELISA (MBL/eBioscience and R&D systems respectively).
IRF8 Phosphorylation
RAW LXRα WT cells were treated with GW3965 (1 μ mol/L) for 24 hours. Cells were lysed in phospho-lysis buffer as in56. Lysates were incubated with antibodies against IRF8 (Santa Cruz, sc-13043x) overnight at 4 °C. Protein complexes containing IRF8 were immunoprecipitated and analysed as in56. Antibodies against phospho-Tyrosine coupled to HRP (Merck-Millipore, 4G10), IRF8 (Santa Cruz, sc-6058x) or Hsp90 were used.
Chromatin Immunoprecipitation
ChIP experiments were performed and analysed as described30,56 except cells were cross-linked with 1.5 mmol/L of ethylene glycol-bis (succinimidylsuccinate) for 20 min, followed by 10 min with 1% formaldehyde solution. The antibodies employed include: LXR60, IRF8 (sc-6058x) and rabbit IgG (Cell signalling, 2729S). The IL-18BP, SREBP1c or β -actin promoters were amplified with primers shown in Supplementary Table 1.
Statistical analysis
Data is presented as mean ± SEM when multiple independent experiments (carried out in independent BMDM preparations from different mice) are considered. Data from a representative experiment (of 2 or 3) using an individual BMDM preparation is presented as mean ± SD of triplicate measurements. Differences were considered significant at p < 0.05 using a two-tailed Student t-test.
Additional Information
How to cite this article: Pourcet, B. et al. The nuclear receptor LXR modulates interleukin-18 levels in macrophages through multiple mechanisms. Sci. Rep. 6, 25481; doi: 10.1038/srep25481 (2016).
References
Dinarello, C. A., Novick, D., Kim, S. & Kaplanski, G. Interleukin-18 and IL-18 Binding Protein. Front Immunol 4, 289 (2013).
Unlu, S. et al. Phosphorylation of IRF8 in a pre-associated complex with Spi-1/PU.1 and non-phosphorylated Stat1 is critical for LPS induction of the IL1B gene. Mol Immunol 44, 3364–3379 (2007).
Puren, A. J., Fantuzzi, G. & Dinarello, C. A. Gene expression, synthesis, and secretion of interleukin 18 and interleukin 1beta are differentially regulated in human blood mononuclear cells and mouse spleen cells. Proc Natl Acad Sci USA 96, 2256–2261 (1999).
Kim, Y. M., Im, J. Y., Han, S. H., Kang, H. S. & Choi, I. IFN-gamma up-regulates IL-18 gene expression via IFN consensus sequence-binding protein and activator protein-1 elements in macrophages. J Immunol 165, 3198–3205 (2000).
Kim, Y. M. et al. Roles of IFN consensus sequence binding protein and PU.1 in regulating IL-18 gene expression. J Immunol 163, 2000–2007 (1999).
Gracie, J. A., Robertson, S. E. & McInnes, I. B. Interleukin-18. J Leukoc Biol 73, 213–224 (2003).
Suk, K., Yeou Kim, S. & Kim, H. Regulation of IL-18 production by IFN gamma and PGE2 in mouse microglial cells: involvement of NF-kB pathway in the regulatory processes. Immunol Lett 77, 79–85 (2001).
Ghayur, T. et al. Caspase-1 processes IFN-gamma-inducing factor and regulates LPS-induced IFN-gamma production. Nature 386, 619–623 (1997).
Gu, Y. et al. Activation of interferon-gamma inducing factor mediated by interleukin-1beta converting enzyme. Science 275, 206–209 (1997).
Mariathasan, S. et al. Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature 430, 213–218 (2004).
Mariathasan, S. et al. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 440, 228–232 (2006).
Ozaki, E., Campbell, M. & Doyle, S. L. Targeting the NLRP3 inflammasome in chronic inflammatory diseases: current perspectives. J Inflamm Res 8, 15–27 (2015).
Druilhe, A., Srinivasula, S. M., Razmara, M., Ahmad, M. & Alnemri, E. S. Regulation of IL-1beta generation by Pseudo-ICE and ICEBERG, two dominant negative caspase recruitment domain proteins. Cell Death Differ 8, 649–657 (2001).
Saleh, M. et al. Enhanced bacterial clearance and sepsis resistance in caspase-12-deficient mice. Nature 440, 1064–1068 (2006).
Saleh, M. et al. Differential modulation of endotoxin responsiveness by human caspase-12 polymorphisms. Nature 429, 75–79 (2004).
Lamkanfi, M. et al. INCA, a novel human caspase recruitment domain protein that inhibits interleukin-1beta generation. J Biol Chem 279, 51729–51738 (2004).
Hurgin, V., Novick, D. & Rubinstein, M. The promoter of IL-18 binding protein: activation by an IFN-gamma -induced complex of IFN regulatory factor 1 and CCAAT/enhancer binding protein beta. Proc Natl Acad Sci USA 99, 16957–16962 (2002).
Bachmann, M., Paulukat, J., Pfeilschifter, J. & Muhl, H. Molecular mechanisms of IL-18BP regulation in DLD-1 cells: pivotal direct action of the STAT1/GAS axis on the promoter level. J Cell Mol Med 13, 1987–1994 (2009).
Kiss, M., Czimmerer, Z. & Nagy, L. The role of lipid-activated nuclear receptors in shaping macrophage and dendritic cell function: From physiology to pathology. J Allergy Clin Immunol 132, 264–286 (2013).
Schultz, J. R. et al. Role of LXRs in control of lipogenesis. Genes Dev 14, 2831–2838 (2000).
Collins, J. L. et al. Identification of a nonsteroidal liver X receptor agonist through parallel array synthesis of tertiary amines. J Med Chem 45, 1963–1966 (2002).
A.-Gonzalez, N. & Hidalgo, A. Nuclear Receptors and Clearance of Apoptotic Cells: Stimulating the Macrophage’s Appetite. Front Immunol 5, 211 (2014).
Pascual-Garcia, M. & Valledor, A. F. Biological roles of liver X receptors in immune cells. Arch Immunol Ther Exp (Warsz) 60, 235–249 (2012).
Joseph, S., Castrillo, A., Laffitte, B., Mangelsdorf, D. & Tontonoz, P. Reciprocal regulation of inflamation and lipid metabolism by liver X receptors. Nat Med 9, 213–219 (2003).
Steffensen, K. R., Jakobsson, T. & Gustafsson, J. A. Targeting liver X receptors in inflammation. Expert Opin Ther Targets 17, 977–990 (2013).
Ghisletti, S. et al. Parallel SUMOylation-dependent pathways mediate gene- and signal-specific transrepression by LXRs and PPARgamma. Mol Cell 25, 57–70 (2007).
Ito, A. et al. LXRs link metabolism to inflammation through Abca1-dependent regulation of membrane composition and TLR signaling. Elife 4, e08009 (2015).
Valledor, A. F., Xaus, J., Marques, L. & Celada, A. Macrophage colony-stimulating factor induces the expression of mitogen-activated protein kinase phosphatase-1 through a protein kinase C-dependent pathway. J Immunol 163, 2452–2462 (1999).
Wagner, B. L. et al. Promoter-specific roles for liver X receptor/corepressor complexes in the regulation of ABCA1 and SREBP1 gene expression. Mol Cell Biol 23, 5780–5789 (2003).
Pourcet, B. et al. LXRalpha regulates macrophage arginase 1 through PU.1 and interferon regulatory factor 8. Circ Res 109, 492–501 (2011).
Pourcet, B. & Pineda-Torra, I. Transcriptional regulation of macrophage arginase 1 expression and its role in atherosclerosis. Trends Cardiovasc Med 23, 143–152 (2013).
Tamura, T., Thotakura, P., Tanaka, T. S., Ko, M. S. & Ozato, K. Identification of target genes and a unique cis element regulated by IRF-8 in developing macrophages. Blood 106, 1938–1947 (2005).
Liu, L. et al. Lipopolysaccharide activates ERK-PARP-1-RelA pathway and promotes nuclear factor-kappaB transcription in murine macrophages. Hum Immunol 73, 439–447 (2012).
Stoll, S. et al. Production of functional IL-18 by different subtypes of murine and human dendritic cells (DC): DC-derived IL-18 enhances IL-12-dependent Th1 development. Eur J Immunol 28, 3231–3239 (1998).
Zuercher, W. J. et al. Discovery of tertiary sulfonamides as potent liver X receptor antagonists. J Med Chem 53, 3412–3416 (2010).
Haneklaus, M., O’Neill, L. A. & Coll, R. C. Modulatory mechanisms controlling the NLRP3 inflammasome in inflammation: recent developments. Curr Opin Immunol 25, 40–45 (2013).
Hong, C. et al. Constitutive activation of LXR in macrophages regulates metabolic and inflammatory gene expression: identification of ARL7 as a direct target. J Lipid Res 52, 531–539 (2011).
Wang, C. et al. Characterization of murine macrophages from bone marrow, spleen and peritoneum. BMC Immunol 14, 6 (2013).
Badimon, L. Interleukin-18: a potent pro-inflammatory cytokine in atherosclerosis. Cardiovasc Res 96, 172–175; discussion 176–180 (2012).
Waddington, K. E., Jury, E. C. & Pineda-Torra, I. Liver X receptors in immune cell function in humans. Biochem Soc Trans 43, 752–757 (2015).
Birrell, M. A. et al. Novel role for the liver X nuclear receptor in the suppression of lung inflammatory responses. J Biol Chem 282, 31882–31890 (2007).
Higham, A. et al. The role of the liver X receptor in chronic obstructive pulmonary disease. Respir Res 14, 106 (2013).
Latz, E., Xiao, T. S. & Stutz, A. Activation and regulation of the inflammasomes. Nat Rev Immunol 13, 397–411 (2013).
Derangere, V. et al. Liver X receptor beta activation induces pyroptosis of human and murine colon cancer cells. Cell Death Differ 21, 1914–1924 (2014).
Kumar, N. et al. The benzenesulfoamide T0901317 [N-(2,2,2-trifluoroethyl)-N-[4-[2,2,2-trifluoro-1-hydroxy-1-(trifluoromethyl)ethy l]phenyl]-benzenesulfonamide] is a novel retinoic acid receptor-related orphan receptor-alpha/gamma inverse agonist. Mol Pharmacol 77, 228–236 (2010).
Qu, Y. et al. Pannexin-1 is required for ATP release during apoptosis but not for inflammasome activation. J Immunol 186, 6553–6561 (2011).
Sugawara, S. et al. Neutrophil proteinase 3-mediated induction of bioactive IL-18 secretion by human oral epithelial cells. J Immunol 167, 6568–6575 (2001).
Vigano, E. & Mortellaro, A. Caspase-11: the driving factor for noncanonical inflammasomes. Eur J Immunol 43, 2240–2245 (2013).
Tsutsui, H., Matsui, K., Okamura, H. & Nakanishi, K. Pathophysiological roles of interleukin-18 in inflammatory liver diseases. Immunol Rev 174, 192–209 (2000).
Becker, A. M. et al. IRF-8 extinguishes neutrophil production and promotes dendritic cell lineage commitment in both myeloid and lymphoid mouse progenitors. Blood 119, 2003–2012 (2012).
Holtschke, T. et al. Immunodeficiency and chronic myelogenous leukemia-like syndrome in mice with a targeted mutation of the ICSBP gene. Cell 87, 307–317 (1996).
Tsujimura, H. et al. ICSBP/IRF-8 retrovirus transduction rescues dendritic cell development in vitro . Blood 101, 961–969 (2003).
Hambleton, S. et al. IRF8 mutations and human dendritic-cell immunodeficiency. N Engl J Med 365, 127–138 (2011).
Mancino, A. et al. A dual cis-regulatory code links IRF8 to constitutive and inducible gene expression in macrophages. Genes Dev 29, 394–408 (2015).
Becker, C. E. & O’Neill, L. A. Inflammasomes in inflammatory disorders: the role of TLRs and their interactions with NLRs. Semin Immunopathol 29, 239–248, doi: 10.1007/s00281-007-0081-4 (2007).
Torra, I. P. et al. Phosphorylation of liver X receptor alpha selectively regulates target gene expression in macrophages. Mol Cell Biol 28, 2626–2636 (2008).
Pineda-Torra, I., Gage, M., de Juan, A. & Pello, O. M. Isolation, Culture, and Polarization of Murine Bone Marrow-Derived and Peritoneal Macrophages. Methods Mol Biol 1339, 101–109 (2015).
Peet, D. J. et al. Cholesterol and bile acid metabolism are impaired in mice lacking the nuclear oxysterol receptor LXR alpha. Cell 93, 693–704 (1998).
Bories, G. et al. Liver X receptor activation stimulates iron export in human alternative macrophages. Circ Res 113, 1196–1205 (2013).
Pehkonen, P. et al. Genome-wide landscape of liver X receptor chromatin binding and gene regulation in human macrophages. BMC Genomics 13, 50 (2012).
Acknowledgements
We are grateful to Xiaoyu Hu (Hospital for Special Surgery, NYC) for providing the IRF8−/− bone marrow cells and to Liz Jury (University College London) for providing access to human macrophages. GSK1440233 was kindly provided by Dr Veronique Birault (GSK). This work was supported by a Medical Research Council New Investigator Grant G0801278 (IPT), British Heart Foundation Project Grant PG/13/10/30000 (IPT), and Ministerio de Economia y Competitividad (MINECO) Grant SAF2014-56819-R (AC) and SAF2014-57856 (AFV).
Author information
Authors and Affiliations
Contributions
B.P., M.G., T.L., K.W., O.P. and A.C. performed experiments. B.P., M.G., T.L., O.P., A.C. and I.P.-T. analysed data. A.V. and K.R.S. provided additional materials. M.G., B.P. and I.P.-T. wrote the manuscript. I.P.-T. secured funding.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Pourcet, B., Gage, M., León, T. et al. The nuclear receptor LXR modulates interleukin-18 levels in macrophages through multiple mechanisms. Sci Rep 6, 25481 (2016). https://doi.org/10.1038/srep25481
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep25481
This article is cited by
-
LXRα promotes cell metastasis by regulating the NLRP3 inflammasome in renal cell carcinoma
Cell Death & Disease (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
