
Abstract
AgI nanoionics-based resistive switching memories were studied in respect to chemical stability of the Ag/AgI interface using x-ray absorption spectroscopy. The apparent dissolution of Ag films of thickness below some tens of nanometers and the loss of electrode/electrolyte contact was critically addressed. The results evidently show that there are no chemical interactions at the interface despite the high ionic mobility of Ag ions. Simulation results further show that Ag metal clusters can form in the AgI layer with intermediate-range order at least up to next-next nearest neighbors, suggesting that Ag can permeate into the AgI only in an aggregated form of metal crystallite.
Similar content being viewed by others


Introduction
Redox-based resistive switching memories (ReRAM) are current major candidate for replacing the state-of-the-art dynamic random access memory and non-volatile flash memories in future nanoelectronics and information technology1 and new encouraging concept for logic2 and neuromorphic3 applications are feasible today. Among different types of switching mechanisms the electrochemical metallization memories (ECM) demonstrate advantages of potential multi-bit storage4, nanosecond-ranged switching time5 and low power consumption6. The structure of the memory cell is simple to fabricate and consists of two electrodes with solid electrolyte in-between; the active (oxidizable) electrode is typically Ag or Cu and an inert (not dissolvable) material such as Pt, Ir, W or TiN are used as auxiliary electrode. For solid electrolyte material transporting the cations (e.g. Ag+), typically glasses but also crystalline materials are used6,7. The main disadvantage for using glasses or non-stoichiometric materials with excess metallic/non-metallic components is that the margin of transport properties is largely determined by the particular chemical composition. As typical example can be considered the system Ag/GeSex, where not only the total conductivity changes by orders of magnitude but also the electronic and ionic transference numbers vary substantially depending on the amount of dissolved Ag8. An additional complication appears when the active electrode material dissolves chemically into the electrolyte e.g. Ag in GeSx9. On one hand side the metal dissolution changes the transport properties of the host material and on the other hand side this dissolution may disturb the mechanical (and electric) contact between the active electrode and the electrolyte. To avoid such type of complications, materials with almost minimum possibility for deviation from the stoichiometry such as RbAg4I510 or AgI11,12 have been studied recently. However, even for AgI thin films we have observed that thin layer of Ag apparently dissolves into AgI resulting in almost complete loss of electric contact, which has not been reported for bulk AgI yet.
Therefore, it is important to investigate the atomic exchange and chemical interaction between the active electrode and the solid electrolyte. However, Ag/AgI is generally a “soft” system that can easily change its chemistry or structure under physical or electrochemical stresses. Thus, analyses using conventional chemical analysis technique such as photoemission spectroscopy or Auger electron spectroscopy are quite challenging because the surface etching process which is inevitable due to its short probing depth, can deteriorate the soft cells. In case of Auger electron spectroscopy or electron energy loss spectroscopy, the electron beam donates charges onto the sample again changing the stoichiometry. Therefore, a non-destructive way to examine the chemistry of the Ag/AgI system is required. Moreover, in the Ag/AgI system, it is in principle impossible to distinguish by valence states, the metallic Ag atoms of the top electrode from those diffused/ditributed into the AgI electrolyte itself. Hence, structural information of the specific Ag atoms is further needed to clarify the correlation between the chemistry and the site of the Ag atoms.
This work presents results demonstrating loss of electric contact in nano-crossbar structured ECM memory cells with down-to-30 nm-thick Ag active electrodes. We show that the X-ray absorption spectroscopy (XAS) can reveal both structural and chemical information of the Ag ions; the chemical shift in energy of the main peak reflects the chemistry while the X-ray absorption fine structure (XAFS) does the local structure of the photon-absorbing atoms at the same time. The chemistry of the Ag and I ions which are supposedly buried under the additional Ag, can be analysed in a non-destructive way due to the long penetration depth (~ a few microns) and low momentum of photons. The evolution in the XAS spectra upon the additional Ag deposition was particularly investigated in order to elucidate the analysis.
Results
Figure 1(a) shows normalized Ag L3-edge XAS of the 50 nm-thick bare AgI film and the AgI films on which 10, 20 and 50 nm-thick Ag metal is deposited additionally. Ag L3-edge XAS reflects the unoccupied s or d orbital states with creation of a core hole at the 2p3/2 level. Since AgI and Ag metal have nominally 4d105s0 and 4d105s1 configurations respectively, the near-edge features show mostly ns states with n ≥ 5. It is clearly observed that the overall spectrum changes according to the Ag deposition, reflecting the incorporation of the Ag metal in the film. For comparison, a spectrum of an Ag foil is appended. As the thickness of the additional Ag increases, the XAFS oscillation of the AgI film becomes similar to that of the Ag metal foil systematically. If there is no chemical reaction between the AgI film and the Ag metal layer, the features could be separable into the respective XAFS features. The contribution of the Ag metal would be approximately 4 times larger compared to the same amount of AgI because of the higher packing density as well as the full atomic fraction. This resulted in a substantial resemblance in the XAFS features of all the Ag deposited film to the Ag foil as shown by the vertical lines with negligible contribution of the AgI itself (asterisk).

(a) Ag L3-edge XANES and (b) their derivative spectra of the AgI films after Ag deposition with thicknesses of 0, 10, 20 and 50 nm. The spectrum of Ag foil is appended for comparison.
Generally once a core hole is created in the XAS process, the effective positive charge of the core hole is screened by the valence electrons with a different degree depending on the number of the valence electrons. The edge energy in the XAS spectra, which can be estimated by the maximal slope of the lowest-energy feature, therefore reflects the chemical valence of the photo-excited atom. The derivative spectra of the samples are shown in Fig. 1(b). The peak maxima in the derivative spectra at a photon energy of 3551 eV reflect the Ag0 ions as in the Ag metal, while those at 3552 eV should be referred to the Ag+1 ions as in AgI (shown by vertical lines). The intensity of the Ag0 (Ag+1) feature increases (decreases) as the thickness of the additional Ag increases, again reflecting the decrease in the average value of the Ag valence from +1 to 0. In fact, the highest peak in the spectrum of AgI originates from the d orbital contribution due to slightly incomplete occupation of 4d shell in Ag+ ions13, while Ag metal has no d orbital contribution due to completely filled 4d shell. Since the energy of the unoccupied 4d shell is lower than that of the 5s shell, the amount of energy shift due to empty 5s shell becomes small apparently (~−1 eV).
It might be inferred from Fig. 1 that the addition of Ag seemingly forms an upper layer without having chemical interaction with the AgI film. However, we have found that such upper layer on AgI is quite unstable and has a high resistance even up to a thickness of a few tens of nanometers. This suggests that the Ag metal can easily penetrate into the AgI film. Although the material of interests in this work is not merely superionic conductor14 but a low temperature modification, γ-AgI, Ag+ ions are shown to be the mobile species with a transference number of 0.8 approximately (while the I− ions hardly move). Thus, the added/dissolved Ag atoms could possibly intercalate inside the AgI domains without having substantial chemical interactions. Such intercalation could increase the number of reduced Ag ions within the AgI film during the cell operation and cycling. It has been suggested the origin of resistive switching behavior in AgI cell is the formation of filamentary network of reduced Ag ions11 demonstrating quantified conductivity12. Therefore, examination of the presence of reduced Ag inside the AgI film is very important to understand the electrical property of the electrolytic AgI cell. Up to this point, however, it is still unclear whether the incorporated Ag interacts chemically with the AgI or not, because the near-edge spectra in Fig. 1 reflect the averaged electronic structures over all the Ag atoms regardless of their sites- either in the Ag metal or in the AgI domains. It will be shown in the following figures that chemical interactions occur neither in the diffused Ag metal nor in the AgI domains.
Figure 2(a) shows the difference between the XAS spectra. The difference spectrum obtained by subtracting the AgI spectrum from the (10 nm Ag) + AgI film show the unoccupied electronic structure of added Ag separately out of the contribution of the AgI because the thicknesses of the AgI are identical as depicted in Fig. 2(b). Likewise, the difference spectrum of [(20 nm Ag)+AgI] – [(10 nm Ag)+AgI] and that of [(50 nm Ag)+AgI] – [(20 nm Ag) + AgI] show selectively the electronic structure of Ag added by additional 10 nm-thick Ag evaporation and by further 30 nm-thick Ag evaporation, respectively. The similarity in overall XAFS features as well as the edge energies in all the difference spectra (e.g. shown by vertical lines) to the XAS spectrum of Ag foil, evidently shows that the added Ag aggregated in a face-centered-cubic (fcc) structure as in Ag metal. This suggests that there is no intercalation of single Ag0 ion in the AgI layer, because otherwise the local structure of Ag ions should have changed according to the zinc-blend structure of AgI. Therefore, it is confirmed that the diffused Ag remains as metallic Ag0.

(a) The difference spectra of the Ag+AgI films taken by subtracting the spectrum of the film of thinner Ag. The exact coincidence with the spectrum of metallic Ag in regard of edge energy and XAFS oscillation strongly suggests absence of any chemical reaction at the Ag/AgI interfaces. (b) Schematic description to account for the difference analysis.
Figure 3 shows the I L3-edge XAS spectra of the AgI films with and without Ag evaporation. Because the I− ions have the electron configuration almost filled (5s2p6), the near-edge features reflect primarily the 6s unoccupied states which are hybridized with the Ag 4d or 5s states. Hence Fig. 3 reflects the chemistry of the Ag+ ions in the AgI domains selectively as well. Overall similarity in the XAFS features reflects that the local structures of the I− ions in the AgI films are invariant regardless of the addition of Ag. The identical near-edge features further show that the Ag orbital states that hybridize with I orbitals have the same electron configurations implying that the chemistry of Ag ions in AgI does not change upon the Ag addition. Combined with the results in Fig. 2, it can be concluded that the local structures of both Ag metal and the AgI film remain unchanged despite the penetration of the Ag metal into the AgI film.

I L3-edge XANES.
No substantial difference between the spectra is observed suggesting the solid I-Ag/I bond structures in AgI.
Discussion
It is shown in the previous section that the chemistry and local structures in the AgI domains are not altered by the Ag addition. Such stability in chemistry of the AgI originates from the complete filling of I 5p shell; added Ag cannot donate electrons to the completely filled I 5p orbitals. Furthermore, we do not observe any charge transfer between added Ag and the Ag+ ions in AgI; it was shown in Fig. 2 that the added Ag has the same chemistry and local structure with Ag metal, while it was shown in Fig. 3 that the electron configuration of the Ag+ ions in AgI do not vary under the Ag addition.
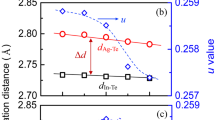
Although we cannot expect some structural changes in the atomic scale, the penetration of Ag into AgI can take effect in a scale of several tens of atoms instead. The crystallinity of the AgI films after the Ag deposition was examined using x-ray diffraction. Figure 4 shows the θ–2θ diffraction patterns of the same samples in a semi-logarithmic plot. The peaks near 2θ = 23.65° show the γ-AgI (111) diffraction while the small features near 2θ = 25.7° show the features of β-AgI15,16, whose concentration is below 1% of the majority (γ-AgI). The features from the Ag metal (fcc) are also observed. Their intensities increase with the amount of added Ag from the obvious reason. In the inset of Fig. 4, the γ-AgI (111) diffraction peaks are magnified. It is clearly shown that the intensity decreases with increasing Ag thickness. This systematic decrease in intensity cannot originate from an experimental artifact such as the anomalous absorption of the diffracted beam by Ag overlayers because the intense Si (400) diffractions from the Si substrate shown in the right-hand side, which should be also affected, are almost constant of Ag thickness. Thus, the decrease of the γ-AgI (111) diffraction peaks definitely reflects some disorder effect in the AgI films17. The tiny features of forbidden Si (200) diffraction appear to evolve somehow systematically; plausibly, it could originate from the minute differences in the measurement geometry of the mounted samples and x-ray scattering plane, which can hardly influence the intensities of the other “allowed” diffractions.

XRD of the (Ag+)AgI films showing the similar crystal structure of the γ-AgI phase except for the increasing features of metallic Ag layer.
Inset: a closer look on the AgI (111) peaks showing that the variation of intensity and width (table) with increasing Ag thickness.
The full-width-at-half-maximum (FWHM) of the (111) peaks are tabulated in the figure. Although the FWHM values are contributed by the peak split due to X-ray satellite (~0.06° at 2θ = 23.65°), they show a clear tendency on the Ag thickness. It is shown that the FWHM of the 10 nm Ag + AgI film is slightly smaller than that of the pure AgI film. This suggests that some possible bombardment effects during the Ag deposition would rather improve the crystallinity of the films. Then the increased FWHMs as well as the decreased intensities in the case of 20 nm Ag + AgI and 50 nm Ag + AgI films should be attributed to the weakened long-range orders in the AgI films.
The long-range order can be partially broken due to the permeability of the Ag ions. It apparently contradicts to the results of this work which strongly suggest the constant local structures in Ag/AgI. However, the apparent contradiction can be resolved if we consider a case that the breaking of the long range order is induced by a large scale intervention of Ag metal clusters in the AgI matrix. The simulation results for the Ag L3-edge XAS spectrum using an ab-initio full multiple scattering (FMS) code, FEFF818, is shown in Fig. 5. The simulation was performed based on the fcc structure of bulk metal confining the range of structural orders up to a radius of RFMS. The simulated spectra depict that the main features shown by the dash lines in Figs. 1 and 2 are reproduced only when the RFMS values exceed 5.01 Å, which corresponds to the distance between the next-next nearest neighbors. The number of atoms in the metallic cluster is estimated to be sufficiently large (43 at least) if assumed a spherical symmetry. This indicates that the Ag metal crystallite in the AgI should consist of at least a few tens of Ag atoms. Due to this large number, the Ag atoms in the metallic cluster can become almost chemically inactive and structurally ordered as in the bulk metal even though they are embedded in the AgI layers.

Theoretical XANES of Ag metal crystallites calculated with considering full multiple scatterings within a radius of RFMS.
Inset: Schematic diagram for possible Ag penetrations into AgI in a sub-mesoscopic scale.
The inset in Fig. 5 depicts schematically the possible atomic configurations in a sub-mesoscopic scale. The aggregated Ag0 ions might form separated clusters or a protrusion of the topmost Ag layer toward the AgI. Because of the large-scale aggregation of the Ag metal, the electrical conductance of the topmost Ag layer would be much lower than the case of Ag metal unless the metallic clusters are well connected together with sufficient amount of additional Ag. The low conductance in the case of insufficient thickness of active electrode is indeed observed for 30 nm Ag + AgI system. Figure 6 shows the results of electrical I–V sweep measurements for the Pt (100 nm)/AgI (30 nm)/Ag crossbar cells with a 30 nm Ag and for comparison sufficiently thick (~200 nm) Ag layers. The AgI cell with thick electrodes exhibits typical ECM operation, while the 30 nm Ag + AgI cell shows no resistance switching but remains in a very high resistance state (> 1 GΩ) even under an excessive applied voltage (> 2 V). The high resistance should be attributed to the loss of contact of the Ag electrode to AgI or between the Ag clusters, suggesting substantial dissolution of Ag into AgI consistent to the findings in this work. Such separated and inactive metal crystallites should interrupt the periodic atomic arrangement in the AgI resulting in a weakened long-range order in the AgI film (Fig. 4) without loss of local structural orders (Fig. 3). We expect for future work it would be quite meaningful to verify the sub-mesoscopic structure of the Ag/AgI system with a non-destructive microscopy, such as the transmission x-ray microscopy.

Current-voltage sweeps recorded by the first cycles in Pt (100 nm)/AgI (30 nm)/Ag [200 nm (solid curve) or 30 nm (dotted)] micro-crossbars.
The sweep rates were 20 mV/s and 2 V/s for the 200 nm Ag and 30 nm Ag samples, respectively. The arrows show the directions of the sweeps.
In summary, we utilized both the near-edge feature analysis and the fine structure analysis to investigate the chemistry and local structure in the Ag metal + AgI interface structures. We did not find signature of chemical interaction between Ag and AgI or any local structural deformation under the Ag addition. Weakened long-range order in AgI suggests the formation of Ag metal crystallites in the AgI film in a sub-mesoscopic scale.
Methods
Sample Preparation and Characterization. The 50 nm-thick AgI films (Alfa Aesar, 99.9% metals basis) were prepared on SiO2 (450 nm)/Si substrates by thermal evaporation at room temperature. Ag films with thicknesses of 10, 20 and 50 nm were deposited additionally by electron beam evaporation. Details on the micro-crossbar fabrication as well as the electrical measurement (Fig. 6) can be found in Ref. 12. X-ray diffraction was measured using X'Pert Pro diffractometer (PANalytical) equipped with Cu Kα source. The diffraction patterns show that all the AgI films are mostly in γ-phase with different degrees of long range orders (Fig. 4).
X-ray absorption spectroscopy. The XAS at the Ag L3- and I L3-edges were performed at the A1 beamline in DORIS III, Deutsches Elektronen-Synchrotron (DESY) in Germany. The Lα1,2 fluorescence was measured using a silicon drift detector equipped in a rectangular position with respect to the incident x-ray beam. The thin film samples were rotated by 45° (with respect to the incident x-ray) to circumvent any issues on polarization dependence.
References
Waser, R. & Aono, M. Nanoionics-based resistive switching memories. Nat. Mater. 6, 833–840 (2007).
Borghetti, J., Snider, G. S., Kuekes, P. J., Yang, J. J., Stewart, D. R. & Williams, R. S. ‘Memristive’ switches enable ‘stateful’ logic operations via material implication. Nature. 464, 873–876 (2010).
Ohno, T., Hasegawa, T., Tsuruoka, T., Terabe, K., Gimzewski, J. K. & Aono, M. Short-term plasticity and long-term potentiation mimicked in single inorganic synapses. Nat. Mater. 10, 591–595 (2011).
Fujisaki, Y. Overview of emerging semiconductor non-volatile memories. IEICE Electron. Express 9, 908–925 (2012).
Kügeler, C. et al. Fast resistive switching in WO3 thin films for non-volatile memory applications. Proceedings of the 9th IEEE Conference on Nanotechnology. 900–903 (2009).
Valov, I., Waser, R., Jameson, J. R. & Kozicki, M. N. Electrochemical metallization memories-fundamentals, applications, prospects. Nanotechnology. 22, 254003 (2011).
Hasegawa, T., Terabe, K., Tsuruoka, T. & Aono, M. Atomic Switch: Atom/Ion Movement Controlled Devices for Beyond Von-Neumann Computers. Adv. Mater. 24, 252–267 (2012).
Kawasaki, M., Kawamura, J., Nakamura, Y. & Aniya, M. Ionic conductivity of Agx(GeSe3)1−x (0< x 0.571) glasses. Solid State Ionics. 123, 259–269 (1999).
Cho, D.-Y., Valov, I., van den Hurk, J., Tappertzhofen, S. & Waser, R. Direct Observation of Charge Transfer in Solid Electrolyte for Electrochemical Metallization Memory. Advanced Materials. 24, 4552–4556 (2012).
Valov, I. et al. Atomically controlled electrochemical nucleation at superionic solid electrolyte surfaces. Nat. Mater. 11, 530–535 (2012).
Liang, X. F., Chen, Y., Shi, L., Lin, J., Yin, J. & Liu, Z. G. Resistive switching and memory effects of AgI thin film. J. Phys. D Appl. Phys. 40, 4767–4770 (2007).
Tappertzhofen, S., Valov, I. & Waser, R. Quantum conductance and switching kinetics of AgI based microcrossbar cells. Nanotechnology. 23, 145703 (2012).
Kleppmann, W. G. & Weber, W. Theory of the quadrupolar deformability in silver halides. Physical Review B. 20, 1669–1677 (1979).
Sanson, A., Rocca, F., Armellini, C., Dalba, G., Fornasini, P. & Grisenti, R. Correlation Between I-Ag Distance and Ionic Conductivity in AgI Fast-Ion-Conducting Glasses. Phys. Rev. Lett. 101, 155901 (2008).
Lee, J., Adams, S. & Maier, J. Defect chemistry and transport characteristics of β-AgI. J. Phys. Chem. Sol. 61, 1607–1622 (2000).
Angeretti, S. M., Dalba, G., Fornasini, P., Benfatto, M. & Rocca, F. L1 and L3 x-ray-absorption edges of iodine in AgI studied by multiple-scattering theory using complex potentials. Physical Review B. 44, 11569–11577 (1991).
Dalba, G., Fornasini, P., Rocca, F. & Mobilio, S. Correlation effects in the extended x-ray-absorption fine-structure Debye-Waller factors of AgI. Physical Review B. 41, 9668–9675 (1990).
Ankudinov, A. L., Ravel, B., Rehr, J. J. & Conradson, S. D. Real-space multiple-scattering calculation and interpretation of x-ray-absorption near-edge structure. Phys. Rev. B. 58, 7565–7576 (1998).
Acknowledgements
The authors thank P. Roegels for preparing the thermal evaporation of AgI films and R. Chernikov for assistance in the beamline experiment. We acknowledge the partial support by the Samsung Global Research Outreach Program. The synchrotron experiments were carried out at the A1 beamline in the light source DORIS III at DESY, a member of the Helmholtz Association (HGF). The research of D.-Y.C. is supported by the Alexander von Humboldt fellowship.
Author information
Authors and Affiliations
Contributions
D.-Y.C., I.V., S.T. conceived and designed the experiment. S.T. prepared the samples and D.-Y.C. conducted the experiment. R.W. supervised the research. All authors discussed the results and contributed to writing the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Cho, DY., Tappertzhofen, S., Waser, R. et al. Chemically-inactive interfaces in thin film Ag/AgI systems for resistive switching memories. Sci Rep 3, 1169 (2013). https://doi.org/10.1038/srep01169
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep01169
This article is cited by
-
The Influence Preparation Way on Properties Powders AgI- SiO2
Silicon (2022)
-
Briefly iodized Ag foils: microstructure, structure, optical response and potential as iodine detectors
Applied Physics A (2015)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
