
Abstract
FAAH-like anandamide transporter (FLAT) regulates anandamide transport for hydrolysis and may be an attractive drug target for pain regulation. We aimed to discover potential FLAT antagonists from traditional Chinese medicine (TCM) using virtual screening, ligand-based drug design and molecular dynamics simulation (MD). Guineensine and Retrofractamide A exhibited high Dock Scores in FLAT. Consensus from multiple linear regression (MLR; R2 = 08973) and support vector machine (SVM; R2 = 0.7988) showed similar bioactivities for Guineensine and the FAAH-1 inhibitor (9Z)-1-(5-pyridin-2-yl-1,3,4-oxadiazol-2-yl)octadec-9-en-1-one. Contour of Guineensine to CoMFA and CoMSIA features also imply bioactivity. MD revealed shake or vibration in the secondary structure of FLAT complexed with Guineensine and (9Z)-1-(5-pyridin-2-yl-1,3,4-oxadiazol-2-yl)octadec-9-en-1-one. Ligand movement might contribute to protein changes leading to vibration patterns. Violent vibrations leading to an overall decrease in FLAT function could be the underlying mechanism for Guineensine. Here we suggest Guineensine as a drug-like compound with potential application in relieving neuropathic pain by inhibiting FLAT.
Similar content being viewed by others


Introduction
Neuropathic pain is a multifactor neurogenic disorder caused by physical damage of neurons, cancer and other diseases1. Patients of neuropathic pain suffer from chronic pain as well as mental illnesses such as depression, anxiety and sleeping disorders2. To date, the mechanism of neuropathic pain remains unclear, making diagnosis and treatment difficult1,3,4.
Anandamide is an endogenous cannabinoid formed by the N-acyl-phosphatidylethanolamine-selective phosphodiesterase (NAPE-PLD) catalyzed hydrolysis of N-arachidonoyl-phosphatidyl-ethanolamine (NAPE)5 and has important physiological roles in pain regulation6. However, activity period of anandamide is short due to the rapid inactivation of anandamide by fatty acid amide hydrolyase (FAAH-1)7,8. Catoblism of anandamide is associated with many different diseases, including cancer, cardiovascular disease, obesity and particularly neuropathic pain9,10,11,12. One emerging approach in controlling pain is the modulation of anandamide degradation by targeting FAAH-113,14,15. Several antagonists of FAAH have been successfully developed16,17,18. Recent findings suggest FAAH-1 cytosolic variant FAAH-like anandamide transporter (FLAT)19 as a possible target for regulating pain. Decreased transportation of anandamide to FAAH-1 by inhibiting FLAT may be an alternative to direct antagonism of FAAH.
In this study, we screen for drug-like compound against FLAT from TCM Database@Taiwan20. Ligand based drug design methods were employed to predict bioactivity of the selected ligands. Molecular dynamics were employed to investigate underlying molecular mechanisms that may contribute to FLAT inhibition.
Results
Homology modeling and molecular docking
Suitability of rat proteins as templates for modeling human proteins was assessed by sequence alignment. Alignment of native rat FAAH-1 and native human FAAH-1 sequences showed 79.7% identity and 89.8% similarity. Re-alignment following removal of α2-helices (T9-T76) (termed FLAT sequences for clarification purposes) increased sequence identity and similarity to 86.1% and 95.6%, respectively (Figure 3). We proceeded to model human FLAT structure using rat FLAT structure based on the high sequence identity and similarity of the FLAT sequences. Structural correctness of the modeled human FLAT structure was evaluated with the Ramachandran plot. A total of 491 residues (98.2%) were distributed in the favored region (Figure 4). Table 1 lists the nine residues (1.8%) distributed in the allowed region. Results of the Ramachandran plot suggest that the modeled human FLAT structure is correct.

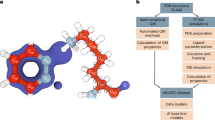
Experimental procedure and structural basis of FLAT simulation.
(a) Simplified scheme of experimental procedures. (b) Structural basis for FLAT structure simulation using FAAH-1. The α2-interacting loop (K255-L278; red) is the binding site opening loop and the helices (P411-N435) colored in cyan are regions in FAAH-1 that interact with the membrane. Presence of the α2-helix (T9-T76; orange) in FAAH-1 was the primary structural difference from FLAT. Human FLAT was modeled from rat FLAT structure, which was computationally prepared by deleting the α2-helix region (amino acids T9-T76) in rat FAAH-1.

Cartoon representation of the anandamide binding site and docking poses of TCM candidates within the binding site.
(a) Enlarged view of the docking site (green) within the modeled human FLAT protein. The front and back sides of the binding site are depicted in red and purple, respectively. (b) Front view of docking site with docked ligands. (9Z)-1-(5-pyridin-2-yl-1,3,4-oxadiazol-2-yl)octadec-9-en-1-one (control), Guineensine and Retrofractamide A are shown in orange, blue and green, respectively. Ser217 and Ile238 are amino acids found adjacent to the binding site. (c) Side view of docking site with docked ligands (45 degrees relative to b).

Sequence alignment of target human FLAT sequence with template rat FLAT sequence.
FLAT sequences refer to that of rat FAAH-1 (PDB: 3K84) and human FAAH-1 (SwissProt: O00519) in which amino acids T9-T76 have been removed. Sequence identity and similarity were 86.1% and 95.6%, respectively.

Ramachandran validation of the modeled human FLAT structure.
A total of 98.2% of the residues were distributed in the favored region. The remaining 1.8% were located in the allowed region. No residues have psi or phi angles in the disfavored regions.
Structure scaffolds of the control and TCM candidates are shown in Figure 5. Candidate selection was primarily based on structural similarities to the control and Dock Score which considers the internal energies of ligands and their corresponding protein-ligand interaction energy. Guineensine37 and Retrofractamide A38, compounds originating from Piper longum39,40 have Dock Scores of 61.45 and 54.01, respectively and were selected as candidates (Table 2). Front and side views of the docking poses of the TCM candidates and the control at the modeled anandamide binding site are illustrated in Figure 2b and 2c. Ligand-protein interactions can be extrapolated from Figure 6. For the control, 16 hydrophobic contacts were formed with 14 residues in FLAT (Figure 6a). Hydrogen bonds (H-bonds) were formed with Ser218, Ile239, Gly240 and Ser242. Guineensine was stabilized by solely hydrophobic interactions (Figure 6b). Similar to the control, Retrofractamide A formed both H-bonds and hydrophobic interactions with FLAT. Since Guineensine and Retrofractamide A primarily differ in the number of carbons in the hydrophobic tail (Figure 5b), the difference in hydrophobic tail length may contribute to differences in scoring functions (Table 2). Based on Ligplot analysis, the O atom located on the hydrocarbon chain was situated to form H-bonds with Met192, Ser194, Ser242 in Retrofractamide A. By contrast, the longer hydrocarbon chain in Guineensine pushes the same O atom away from the H-bond forming amino acids, resulting in Guineensine being stabilized by only hydrophobic interactions (Figure 6b). Cross comparing interaction residues among different test ligands reveals that Met192, Phe193, Ser194, Ile239 and Val492 are residues with which all test ligands formed interactions with (Table 3). The residues functioned to anchor the hydrocarbon tails of the ligands (Figure 7).

Scaffold structures of control compound and TCM candidates.
(a) the alpha-ketoheterocycle inhibitor (9Z)-1-(5-pyridin-2-yl-1,3,4-oxadiazol-2-yl)octadec-9-en-1-one (control) and TCM candidates (b) Guineensine and Retrofractamide A. Guineensine and Retrofractamide A differ in the number of carbons found in the hydrophobic tail region. The number of carbons for Guineensine is depicted in blue and that for Retrofractamide A is depicted in gray.

Protein-ligand interactions analyzed by Ligplot.
(a) (9Z)-1-(5-pyridin-2-yl-1,3,4-oxadiazol-2-yl)octadec-9-en-1-one, (b) Guineensine and (c) Retrofractamide A.

Spatial orientation of key interaction residues with regard to test ligands.
(a) (9Z)-1-(5-pyridin-2-yl-1,3,4-oxadiazol-2-yl)octadec-9-en-1-one (mint), (b) Guineensine (blue) and (c) Retrofractamide A (violet). Common residues are illustrated by yellow surfaces.
Activity prediction using MLR and SVM
Representative descriptors determined by GFA and used for constructing MLR and SVM models were: C_Count, ES_Sum_dsCH, ES_Sum_aasC, ES_Sum_ssNH, ES_Sum_dsN, ES_Sum_aaN, ES_Sum_dO, ES_Sum_sF, AverageBondLength, Num_Chains. Descriptions of each property are listed in Supplementary Table S1. In general, antagonistic activity against FLAT was associated with carbon atom numbers, electrotopolocial state, atom bond length and unbranched chain numbers. The optimum GFA model with an R2 of 0.9403 was:

The coefficient of determination (R2) for the MLR and SVM models constructed using the aforementioned descriptors are 0.8973 (Figure 8a) and 0.7988 (Figure 8b), respectively. Correlation between observed and predicted bioactivity values indicate good prediction accuracy of both models. Predicted pIC50 of the control and TCM candidates using the MLR and SVM models are listed in Table 2. Both models predict bioactivity for the TCM candidates. Similar bioactivities ranging between 7.72–7.89 were predicted for the three compounds with MLR. SVM model predictions show Guineensine and (9Z)-1-(5-pyridin-2-yl-1,3,4-oxadiazol-2-yl)octadec-9-en-1-one to have bioactivities approximately 1.5 logs higher than Retrofractamide A. To reduce possible bias caused by one single model, we used a simplified consensus to summarize our bioactivity prediction results. The criteria for each score given is described in Table 2. The combined results of our MLR and SVM models indicate that Guineensine and the control have similar predicted bioactivities while Retrofractamide A may have lower bioactivities (Table 2). The variation in bioactivity could be related to the number of carbons within the hydrophobic tail since carbon numbers were the primary difference between Guineensine and Retrofractamide A. GFA results in which the number of C atoms is a representative descriptor of bioactivity support this view.

Correlation plot of observed bioactivity verses predicted bioactivities generated by different models.
(a) MLR (b) SVM (c) CoMFA (d) CoMSIA.
Activity prediction using CoMFA and CoMSIA
PLS results of CoMFA and CoMSIA models are tabulated in Table 4. Steric field was the primary determinant of bioactivity in the CoMFA model. At an optimal number of components (ONC) of 6, the cross validation correlation coefficient (q2) of 0.690 and non-cross-validation correlation coefficient (r2) of 0.954 indicated a confident model. CoMSIA models considered multiple factors and the possible models at an ONC of 6 are shown in Table 4. The CoMSIA model integrating steric, hydrophobic and H-bond acceptor properties exhibited the highest cross validation (q2 = 0.802) and non-cross validation (r2 = 0.948) values, thus was selected as the CoMSIA model of choice.
Correlation between the observed and predicted bioactivity values generated by the CoMFA and CoMSIA are shown in Figure 8c and 8d, respectively. Correlation coefficients (R2) of 0.9542 for the CoMFA model and 0.9478 for the CoMSIA model indicated good prediction strength of both models. The range of residuals between predicted and observed value is < ±1.0 (Table 5). Superimposition of aligned ligands with CoMFA feature contours indicate that ligand aromatic heads and bends in the hydrophobic tail region contoured to the steric favoring bulk (yellow) in the CoMFA map (Figure 9a). Additionally, the hydrophobic tail matches the contour disfavoring steric bulk (green). Alignment of the ligands with the bioactivity feature maps suggest activity of the ligands against the target protein FLAT. As expected, the CoMSIA feature map (Figure 9b) differed from CoMFA. Steric favoring contours (yellow), as well as steric disfavoring contours (green), were located near the hydrophobic tail. Both hydrophilic (cyan) and hydrophobic (gray) contours were located near the aromatic head. From the contour of the aromatic head region to the hydrophobic favoring bulk, we speculate that the ligands should exhibit some bioactivity despite the lack of bulk at the steric favoring region. Results from CoMFA and CoMSIA are consistent with results from MLR and SVM predictions and consistently suggest bioactivity of the TCM candidates toward FLAT.

Contour of superimposed ligands to CoMFA and CoMSIA feature maps.
(a) The aromatic head and bend in the hydrophobic tail contoured to the steric favoring region (yellow) and the hydrophobic tail regions contoured to the steric disfavoring region in the CoMFA map. (b) In the CoMSIA map, ligand tail regions contoured to both the steric favoring (yellow) and disfavoring (green) regions. Aromatic heads of the ligands contoured to both the hydrophobic (gray) and hydrophilic (cyan) features. (9Z)-1-(5-pyridin-2-yl-1,3,4-oxadiazol-2-yl)octadec-9-en-1-one, Guineensine and Retrofractamide A (green) are represented in blue, red and green, respectively.
Molecular dynamics (MD) simulation
Molecular dynamics simulations were employed to investigate dynamic stability of the complexes. Characteristics of the protein-ligand complexes are summarized in Table 6. Complexes of the TCM candidates Guineensine and Retrofractamide A had lower total energy levels than the control complex. Globally speaking, binding of TCM candidates did not significantly alter protein characteristics compared to the control FAAH-1 antagonist. On a local scale, calculated RMSD values and total drifts (Table 6) indicate that Retrofractamide A complex was most stable, followed by (9Z)-1-(5-pyridin-2-yl-1,3,4-oxadiazol-2-yl)octadec-9-en-1-one. Guineensine was the least stable of the three complexes.
DSSP analysis allowed visualization of structural changes that contributed to stability differences. Three distinctive patterns, (1) without shake or vibration, (2) shake, or (3) vibration, were observed in the three complexes (Figure 10). The non-vibrating pattern implies a stable secondary protein structure of small changes. The shake pattern refers to isolated peaks of changes whereas the vibration pattern exhibits constant vibrations over a period of time. The shake pattern can be viewed as a pre-vibration signal. As shown in Figure 10a, all three patterns are present in the control complex. Isolated shaking patterns were present from 15–26 ns and accumulated to continuous vibrations from 26–40 ns. Guineensine complex was stable up to 6 ns, but exhibited vibration patterns after 6 ns to the end of MD (Figure 10b). Retrofractamide A complex remained stable during MD, with minor shakes observed in residues 497–577 after 30 ns (Figure 10c). The fluctuations observed here may provide an explanation for the larger RMSDs and drifts recorded in Table 6. Root mean square fluctuation (RMSF) analysis on docking interaction residues provide information on residues that may be important for the protein DSSP changes and ultimately bioactivity against FLAT. Large fluctuations at Ile239, Tyr272 and Gln274 could induce ligand instability and in turn affect the stability of FLAT. (Figure 11, Table 7).

Secondary structure changes observed during the 40 ns MD simulation.
(a) (9Z)-1-(5-pyridin-2-yl-1,3,4-oxadiazol-2-yl)octadec-9-en-1-one, (b) Guineensine and (c) Retrofractamide A. The three complexes demonstrate distinctive change patterns. The “shake” pattern refers to a sudden peak of vibration; the “vibration” pattern refers to observation of continuous vibrations.

Root mean square analysis of FLAT residues implicated in docking.
Yellow, red and blue bars represent data for (9Z)-1-(5-pyridin-2-yl-1,3,4-oxadiazol-2-yl)octadec-9-en-1-one, (b) Guineensine and (c) Retrofractamide A, respectively.
Several analyses were conducted to determine the effects of TCM ligands on FLAT structure with (9Z)-1-(5-pyridin-2-yl-1,3,4-oxadiazol-2-yl)octadec-9-en-1-one as the point of reference (Figure 12). Solvent accessible surface area (SASA) remained relatively constant for protein complexes (Figure 12a) and individual ligands (Figure 12b), indicating no significant changes in protein folding. Average ligand SASAs of control, Guineensine and Retrofractamide A are 6.895 nm/NS2, 6.458 nm/NS2 and 5.488 nm/NS2, respectively, which is most likely related to the hydrophobic tail lengths of each ligand. Changes in complex compactness were evaluated based on radius of gyration (Rg) which measures the mass of atoms relative to the center of mass of the complex. Average and maximum levels of Rg for each complex are tabulate in Table 8. Over the course of time, an increase in Rgcomplex starting at approximately 28 ns was recorded for the control and maximum Rgcomplex of 4.184 nm was reached at 38.46 ns. In Guineensine, changes from the baseline trajectory were observed around 10 ns and peaked to Rgcomplex of 4.7 nm at 18.32 ns. The Rgcomplex for Retrofractamide A was relatively static. In agreement with Rgcomplex results, higher Rgligand, was recorded for Guineensine compared to the other two test ligands (Figure 12d). Intriguingly, Rgcomplex trajectories seemed to be related to pattern changes in Figure 10. Changes in Rgcomplex coincided with shakes or vibrations observed for the control and Guineensine.

Analysis of MD trajectories generated by Gromacs.
Trajectories for (a) protein solvent accessible surface area (SASA), (b) ligand SASA, (c) protein radius of gyration (Rg), (d) ligand Rg, (e) protein mean square deviation (MSD), (f) ligand MSD, (g) protein root mean square deviation (RMSD) and (h) ligand RMSD are shown. Data for (9Z)-1-(5-pyridin-2-yl-1,3,4-oxadiazol-2-yl)octadec-9-en-1-one, guineensine and Retrofractamide A are shown in mint, blue and violet, respectively.
The time dependent movement of atoms from their initial positions (MSD) were in agreement with gyration results. An increase in MSDcomplex (indicated by the increase in slope) was observed for the control (Figure 12e) as the complex becomes more relaxed after 25 ns (Figure 12c). A two-stage change in MSDcomplex was observed for Guineensine (Figure 12e). The steep slope in stage 1 (0–17 ns) matched the expansion of Guineensine complex (Figure 12c) and structure vibration (Figure 10b). During stage 2, the decrease in MSDcomplex slope coincided with increased Guineensine compactness and lower intensity vibrations. Displacement of ligands may also contribute to protein compactness and vibrations. Trajectory of control MSDligand was similar to that for Rgcomplex. For Guineensine, the MSDligand was similar to the trend of its MSDprotein. It is possible that in both cases, ligand shift was a primary force in initiating protein changes recorded in Figure 10 and Figure 12c. The high RMSDprotein of the control as opposed to the TCM candidates (Figure 12g) suggest that complexes formed by the TCM candidates cause less structural perturbation to the FLAT protein. No significant differences in RMSDligand was observed among the three compound (Figure 12h). Distance matrices of the three candidates were also similar (Figure 13).

Distance matrices depicting the smallest distance between residue pairs.
(a) (9Z)-1-(5-pyridin-2-yl-1,3,4-oxadiazol-2-yl)octadec-9-en-1-one, (b) Guineensine and (c) Retrofractamide A.
Discussion
The selected TCM compounds Guineensine and Retrofractamide A are near identical chemical structures with the only difference being the length of the hydrocarbon chain length (Figure 5). The longer tail of Guineensine enables its stabilization by hydrophobic interactions within the FLAT protein (Figure 6). By contrast, the shorter tail in Retrofractamide A formed H-bonds with Met192, Ser194, Ser242 within the hydrophobic interior of the protein. Residues Met192, Phe183, Ser194, Ile239, Val492 were important contact residues that stabilized the tail regions within the docking site. Bioactivity results obtained through QSAR model predictions also suggested an association between bioactivity strength and chain length. Guineensine and the control were both predicted to have higher bioactivities than the shorter Retrofractamide A. MD results further revealed protein secondary structure changes in Guineensine and the control that were not observed in Retrofractamide A. Since ability to initiate unstable vibrations in FLAT showed similar trends to bioactivity (Figure 10), we propose that a possible link might exist between these characteristics. Large fluctuations at Ile239, Tyr272 and Gln274 could contribute to bioactivity by inducing ligand instability, changing protein compactness and affecting the stability of FLAT. Based on the combined results from docking, QSAR predictions and MD simulation, Guineensine exhibits similar patterns to the control and could be a likely candidate for further studies.
The TCM compound Guineensine was predicted to have similar bioactivities with the demonstrated FAAH-1 inhibitor (9Z)-1-(5-pyridin-2-yl-1,3,4-oxadiazol-2-yl)octadec-9-en-1-one. Molecular insights suggest that the inhibitory mechanism may be related to the ability to initiate unstable vibrations in FLAT. Guineensine induced violent vibrations in the FLAT complex. (9Z)-1-(5-pyridin-2-yl-1,3,4-oxadiazol-2-yl)octadec-9-en-1-one induced similar, albeit less extreme, vibration. Gromacs analysis results suggest that ligand movement within the binding site might be associated with protein compactness changes which further translates into global protein structure vibrations. Based on these observations, we suggest that Guineensine may be a possible ligand for inhibiting FLAT transportation of anandamine. These findings may have important implications in designing better alternative for managing neuropathic pain.
Methods
Figure 1a summarizes the overall procedure used in this study.
Homology modeling
The crystal structure of rat fatty-acid amide hydrolase 1 (PDB: 3K84)21 was downloaded from Protein Data Bank. For clarification purposes, all sequence numberings referred to follow those of rat FAAH-1 (PDB: 3K84). The α2 helix (from T9 to T76) of FAAH-1 was computationally removed to form the FLAT structure of rats (Figure 1b). The homology model of human FLAT was constructed by using rat FLAT as the template structure and human FAAH-1 (SwissProt Index: O00519) as the template sequence. RAMPAGE22 was used to verify validity of the predicted structure.
Molecular docking
The modeled human FLAT structure was applied for molecular docking. Over 30,000 compounds were downloaded from TCM Database@Taiwan20 and docked into the anandamide binding site of the modeled human FLAT structure (Figure 2a). All compounds were prepared with CHARMm (Chemistry at HARvard Molecular Mechanics)23 prior to docking to add missing hydrogen atoms. The alpha-ketoheterocycle inhibitor (9Z)-1-(5-pyridin-2-yl-1,3,4-oxadiazol-2-yl)octadec-9-en-1-one in rat FAAH-1 (PDB: 3K84) was used as the control21. Control and TCM compounds were docked using the LigandFit module in Discovery Studio 2.5 (DS 2.5)24. For each protein-ligand complex, five different conformations were generated based on default DS 2.5 settings. Each generated conformation was compared to the conformation of (9Z)-1-(5-pyridin-2-yl-1,3,4-oxadiazol-2-yl)octadec-9-en-1-one within the rat fatty-acid amide hydrolase 1 (PDB:3K84) crystal structure. The conformation most similar to the control in 3K84 and with the highest Dock Score was selected.
Activity prediction using quantitative structure activity relationship (QSAR) Models
A total of 52 ligands25 were selected to calculate molecular properties using the Calculate Molecular Properties protocol packaged in DS 2.5. Genetic function approximation (GFA) was used to identify ten representative descriptors associated with bioactivity. The calculated models were ranked by square correlation coefficient (R2) and the highest R2 model was used to predict bioactivities for the training set and test set. The linear Multiple Linear Regression (MLR) model was constructed with MATLAB26 and the non-liner model utilizing Support Vector Machine (SVM)27,28 was constructed with LibSVM29.
Comparative force field analysis (CoMFA) and comparative similarity indices analysis (CoMSIA) were applied to construct 3D-QSAR models. Ligand alignment was completed using the atom-fit module of SYBYL-X 1.130. In CoMFA, steric and electrostatic fields were calculated using Lennard-Jones potential (LJP) and Coulombic potential, respectively. In CoMSIA, descriptors including steric, hydrogen bond acceptor and donor, hydrophobic and electrostatic fields were calculated by Gaussian functions. Partial least squares (PLS)31 was used for property analysis. Cross validation model was ranked by cross-validated coefficient (q2), whereas non-cross validation was ranked by conventional correlation coefficient (r2), standard error of estimate (SEE) and standard error. The model with the highest q2, r2 values and the lowest SEE and standard error was selected as the optimum model and used to predict bioactivities of the TCM candidates.
Molecular dynamics (MD) simulation
All ligands are prepared by using web server SwissParam (http://swissparam.ch/)32 prior to MD simulation. MD was performed using GROMACS 4.0.733. The force field applied for simulation is detailed elsewhere34. Ligands and human FLAT were combined and immersed into a buffer (or solution) containing cubic box at a buffering distance of 1.2 nm between the complex and the edge of the box. Sodium and chloride ion were added to neutralize complex charge. Complex energies were subsequently minimized with the Steepest Descent method for 5000 steps. The last frame of each energy-minimized structure was used as the initial frame for MD simulation. Electrostatic interactions were calculated with the Particle-Mesh Ewald (PME) method35. The cutoff for PME was 1.0 nm. The time step was set at 2 fs and the number of steps set to 20000000, accumulating to a total MD simulation time of 40 ns.
MD trajectories were analyzed with built-in Gromacs tools. The secondary structure database (DSSP) was installed into Gromacs to analyze protein secondary structure changes36. Program g_energy was used to analyze potential energy, kinetic energy, total energy, temperature, pressure, volume, density, pV and enthalpy changes. Program g_gyrate was used the measure the radius of gyration. Program g_sas was used to compute interaction surface areas between solvent molecules and complexes. Program g_mdmat was used to generate distance matrices which calculate the smallest distance between each residue pairs.
References
Jensen, T. S. et al. A new definition of neuropathic pain. Pain 152, 2204–2205 (2011).
Turk, D. C., Audette, J., Levy, R. M., Mackey, S. C. & Stanos, S. Assessment and treatment of psychosocial comorbidities in patients with neuropathic pain. Mayo Clin Proc 85, S42–50 (2010).
Baron, R., Tolle, T. R., Gockel, U., Brosz, M. & Freynhagen, R. A cross-sectional cohort survey in 2100 patients with painful diabetic neuropathy and postherpetic neuralgia: Differences in demographic data and sensory symptoms. Pain 146, 34–40 (2009).
Mahn, F. et al. Sensory symptom profiles and co-morbidities in painful radiculopathy. PLoS One 6, e18018 (2011).
Liu, J. et al. Multiple pathways involved in the biosynthesis of anandamide. Neuropharmacology 54, 1–7 (2008).
Fride, E. Endocannabinoids in the central nervous system--an overview. Prostaglandins Leukot Essent Fatty Acids 66, 221–233 (2002).
Cravatt, B. F. et al. Supersensitivity to anandamide and enhanced endogenous cannabinoid signaling in mice lacking fatty acid amide hydrolase. Proc Natl Acad Sci U S A 98, 9371–9376 (2001).
McKinney, M. K. & Cravatt, B. F. Structure and function of fatty acid amide hydrolase. Annu Rev Biochem 74, 411–432 (2005).
Hohmann, A. G. & Suplita, R. L. 2nd. Endocannabinoid mechanisms of pain modulation. Aaps J 8, E693–708 (2006).
Jhaveri, M. D., Richardson, D. & Chapman, V. Endocannabinoid metabolism and uptake: novel targets for neuropathic and inflammatory pain. Br J Pharmacol 152, 624–632 (2007).
Lambert, D. M. Allergic contact dermatitis and the endocannabinoid system: from mechanisms to skin care. ChemMedChem 2, 1701–1702 (2007).
Bisogno, T. & Di Marzo, V. Short- and long-term plasticity of the endocannabinoid system in neuropsychiatric and neurological disorders. Pharmacol Res 56, 428–442 (2007).
Karsak, M. et al. Attenuation of allergic contact dermatitis through the endocannabinoid system. Science 316, 1494–1497 (2007).
Maione, S. et al. Analgesic actions of N-arachidonoyl-serotonin, a fatty acid amide hydrolase inhibitor with antagonistic activity at vanilloid TRPV1 receptors. Br J Pharmacol 150, 766–781 (2007).
Jhaveri, M. D., Richardson, D., Kendall, D. A., Barrett, D. A. & Chapman, V. Analgesic effects of fatty acid amide hydrolase inhibition in a rat model of neuropathic pain. J Neurosci 26, 13318–13327 (2006).
Kathuria, S. et al. Modulation of anxiety through blockade of anandamide hydrolysis. Nat Med 9, 76–81 (2003).
Bisogno, T. et al. Arachidonoylserotonin and other novel inhibitors of fatty acid amide hydrolase. Biochem Biophys Res Commun 248, 515–522 (1998).
Zhang, D. et al. Fatty acid amide hydrolase inhibitors display broad selectivity and inhibit multiple carboxylesterases as off-targets. Neuropharmacology 52, 1095–1105 (2007).
Fu, J. et al. A catalytically silent FAAH-1 variant drives anandamide transport in neurons. Nat Neurosci 15, 64–69 (2012).
Chen, C. Y. C. TCM Database@Taiwan: The World's largest traditional Chinese medicine database for drug screening in silico. PLoS One 6, e15939 (2011).
Mileni, M. et al. X-ray crystallographic analysis of alpha-ketoheterocycle inhibitors bound to a humanized variant of fatty acid amide hydrolase. J Med Chem 53, 230–240 (2010).
Lovell, S. C. et al. Structure validation by Calpha geometry: phi,psi and Cbeta deviation. Proteins 50, 437–450 (2003).
Brooks, B. R. et al. CHARMM: the biomolecular simulation program. J Comput Chem 30, 1545–1614 (2009).
Discovery Studio v. 2.5.5. (San Diego, CA).
Sit, S. Y. et al. Oxime carbamate--discovery of a series of novel FAAH inhibitors. Bioorg Med Chem Lett 20, 1272–1277 (2010).
Natick, M., US. MATrix LABoratory. The MathWorks Inc.
Hasegawa, K. & Funatsu, K. Non-Linear Modeling and Chemical Interpretation with Aid of Support Vector Machine and Regression. Curr Comput-Aid Drug 6, 24–36 (2010).
Kim, J. H., Lee, J., Oh, B., Kimm, K. & Koh, I. S. Prediction of phosphorylation sites using SVMs. Bioinformatics 20, 3179–3184 (2004).
Wieland, R. et al. A new library to combine artificial neural networks and support vector machines with statistics and a database engine for application in environmental modeling. Environ Modell Softw 25, 412–420 (2010).
St Louis, M. USA: Tripos. SYBYL-X. 1.1.
Stanton, D. T. QSAR and QSPR Model Interpretation Using Partial Least Squares (PLS) Analysis. Curr Comput-Aid Drug 8, 107–127 (2012).
Zoete, V., Cuendet, M. A., Grosdidier, A. & Michielin, O. SwissParam, a Fast Force Field Generation Tool For Small Organic Molecules. J Comput Chem 32, 2359–2368 (2011).
Hess, B., Kutzner, C., van der Spoel, D. & Lindahl, E. GROMACS 4: Algorithms for highly efficient, load-balanced and scalable molecular simulation. J Chem Theory Comput 4, 435–447 (2008).
Priyakumar, U. D. & MacKerell, A. D. Comparison of the CHARMM27, AMBER4.1 and BMS nucleic acid force fields via free energy calculations of base flipping. Abstr Pap Am Chem S 230, U1391–U1392 (2005).
Darden, T. A. & Pedersen, L. G. Molecular modeling: an experimental tool. Environ Health Perspect 101, 410–412 (1993).
Kabsch, W. & Sander, C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22, 2577–2637 (1983).
Lee, S. A. et al. Methylpiperate derivatives from Piper longum and their inhibition of monoamine oxidase. Arch Pharm Res 31, 679–683 (2008).
Rho, M. C. et al. ACAT inhibition of alkamides identified in the fruits of Piper nigrum. Phytochemistry 68, 899–903 (2007).
Zhang, H., Matsuda, H., Nakamura, S. & Yoshikawa, M. Effects of amide constituents from pepper on adipogenesis in 3T3-L1 cells. Bioorg Med Chem Lett 18, 3272–3277 (2008).
Lin, Z., Liao, Y., Venkatasamy, R., Hider, R. C. & Soumyanath, A. Amides from Piper nigrum L. with dissimilar effects on melanocyte proliferation in-vitro. J Pharm Pharmacol 59, 529–536 (2007).
Acknowledgements
The research was supported by grants from the National Science Council of Taiwan (NSC101-2325-B-039-001), Asia University (100-asia-56, asia100-cmu-2) and China Medical University (DMR-101-094). This study is also supported in part by Taiwan Department of Health Clinical Trial and Research Center of Excellence (DOH101-TD-B-111-004) and Taiwan Department of Health Cancer Research Center of Excellence (DOH101-TD-C-111-005). We are grateful to Asia University and the National Center of High-performance Computing for computer time and facilities.
Author information
Authors and Affiliations
Contributions
The original concept was conceived by C.Y.C. Chen. The study was conducted and analyzed by W.I. Tou. The manuscript was written by W.I. Tou and S.S. Chang. Experimental facilities were provided by C.Y.C. Chen and C.C. Lee.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Tou, W., Chang, SS., Lee, CC. et al. Drug Design for Neuropathic Pain Regulation from Traditional Chinese Medicine. Sci Rep 3, 844 (2013). https://doi.org/10.1038/srep00844
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep00844
This article is cited by
-
Anti-melanogenic activity of Myristica fragrans extract against Aspergillus fumigatus using phenotypic based screening
BMC Complementary Medicine and Therapies (2020)
-
Discovery of Novel Multi-target Inhibitor of angiotensin type 1 receptor and neprilysin inhibitors from Traditional Chinese Medicine
Scientific Reports (2019)
-
Residue-based design of small molecule inhibitor for H1N1, H5N1 and H7N1 mutants
Journal of Molecular Modeling (2016)
-
Susceptible gene of stasis-stagnation constitution from genome-wide association study related to cardiovascular disturbance and possible regulated traditional Chinese medicine
BMC Complementary and Alternative Medicine (2015)
-
First-principles studies of multiferroic and magnetoelectric materials
Science Bulletin (2015)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
