
Abstract
Trichostrongylus colubriformis is a parasitic helminth that primarily infects small ruminants, causing substantial economic losses in the livestock industry. Exploring the microbiome of this helminth might provide insights into the potential influence of its microbial community on the parasite’s survival. We characterised the intestinal microbiome of T. colubriformis that had been collected from the duodenum of sheep, and compared the helminth microbiome with the duodenal microbiome of its host, aiming to identify contributions from the helminth’s environment. At the same time, we explored the isolation of fastidious organisms from the harvested helminth. Primary alpha and beta diversity analyses of bacterial species revealed statistically significant differences between the parasite and the host, in terms of species richness and ecological composition. 16S rRNA differential abundance analysis showed that Mycoplasmoides and Stenotrophomonas were significantly present in T. colubriformis but not in the duodenal microbiome of the sheep. Furthermore, two bacteria, Aeromonas caviae and Aeromonas hydrophila, were isolated from T. colubriformis. Examinations of the genome highlight differences in genome size and profiles of antimicrobial resistance genes. Our results suggest that T. colubriformis carries a specific bacterial community that could be supporting the helminth’s long-term survival in the host’s digestive system.
Similar content being viewed by others


Introduction
Gastrointestinal helminths in farmed livestock, including sheep, cause health and welfare issues and significant economic losses in pasture-based grazing systems1,2. One of the most important helminth species, Trichostrongylus, is zoonotic and is prevalent in pastoral communities around the world where it induces trichostrongyliasis in humans as well as livestock3. In humans, it causes gastrointestinal complications such as hyper-eosinophilia, abdominal pain, diarrhoea and anemia4,5. In sheep, it is found in the small intestine and high levels of infection with Trichostrongylus colubriformis can cause severe enteritis, weight loss, pathological changes in the small intestine that impair nitrogen metabolism, alterations in short-chain fatty acid production, metabolic disorders, and reductions in bone density6,7,8,9,10,11. In infected sheep, there is also a decrease in circulating levels of total protein, albumin, urea, and butyrate, leading to an increase in enteric methane emissions (CH4). These outcomes are associated with alterations in the rumen microbiome, characterized by a combination of suppression of flora responsible for maintaining microbial homeostasis with the promotion of the archaeal community12.
The transcriptome of adult T. colubriformis is linked to peptides directly associated with the host nervous system, digestion of host proteins, or inhibition of host proteases13. Thus, the microbiota and helminths combine to produce metabolites and neurochemicals that have a pivotal role in influencing the segment where the helminths live (the small intestine for T. colubriformis) as well as other compartments of the host’s digestive tract and extra-intestinal tissues14. Most symbionts of helminths are likely to be associated with either the gut or external surfaces, and to be commensals or mutualists contributing principally to host metabolism. An understanding of these organisms, especially any essential bacterial symbionts, might present alternatives to anthelmintics, such as biological control agents1.
Prior research indicates that the richest and most diverse microbial populations occurred in the duodenum of helminth-resistant sheep, suggesting that helminth-resistance involves reorganization of the duodenal microbiome to restrict helminth development15. Another possibility is that the microbiota of the helminth gut represents a target for the development of new strategies for antihelminth treatment and control2. In this study, our main goal was to describe the microbiota of T. colubriformis that were infecting the sheep duodenum, establishing a comparative analysis between the helminth and its host. Simultaneously, we attempted to analyse the presence of fastidious organisms from the harvested T. colubriformis, contributing to our understanding of the microbial ecosystem. This analysis was expected to enhance our understanding of helminth biology, including interactions with the microbiome of the host gut, and suggest potential avenues for developing novel helminth control strategies.
Results
Sequencing description
From randomly selected animals, T. colubriformis and duodenum content were collected for DNA extraction to produce 16S rRNA V3–V4 amplicons. After sequencing the corresponding amplicons, we produced a total of 3,110,866 reads. The paired-end reads were processed including trimmed, filtered, denoised, merged, and chimera removed generating 2,360,424 reads (Supplementary 1). These sequences were converted into 1761 raw ASVs revealing 20 phyla, 36 classes, 69 orders, 126 families, and 184 genera (Supplementary 2).
General 16S rRNA analysis
For microbial populations, the Alpha diversity values, including ACE, Shannon, Chao1, and Observed OTUs index were calculated and compared between the groups (Fig. 1A). These values refer to the abundance and variety of microorganisms present in a particular environment. The results showed statistically significant differences in terms of species richness between the environment determined by ACE and Chao1 index. Based on species diversity represented by Shannon and Observed OTUs no significant differences were found. We also presented the Pearson correlation analysis associated to the Alpha diversity values (Fig. 1B).

Abundance and variety of microorganisms present in duodenum content and T. colubriformis evaluated through Alpha diversity indices, including Observed OTUs, Shannon, Chao1, and ACE. (A) Comparative analysis of Alpha diversity indices employed a Student’s T-test, (B) and Person correlation analysis.
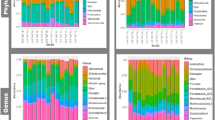
In Fig. 2, the relative abundance of bacteria in both duodenal content and the T. colubriformis parasite is described. T. colubriformis exhibited a substantial presence of the phylum Mycoplasmatota, Pseudomonadota, Bacillota, Campylobacterota, and Actinomycetota (Fig. 2A,B). Particularly the duodenum presented high numbers of the phylum Pseudomonadota, Bacillota, Bacteroidota, and Actinomycetota. Alongside the helminth, at the genus level, the abundant taxa were primarily represented by Escherichia, Mycoplasmoides, Clostridium, and Helicobacter (Fig. 2C,D). In parallel, the duodenum presented high abundance for mainly Escherichia, Clostridium, and Bacteroides.

Relative abundance plot featuring (A) the top 20 phyla, (B) a taxonomy plot categorize by groups presenting phylum classification, (C) the 20 most abundant genera, and (D) the genus taxonomy plot categorized by groups.
Using Principal Component Analysis (PCA) alongside Aitchison distance matrices, we observe two distinct clusters that effectively differentiate T. colubriformis from the duodenal content samples (Fig. 3). The close proximity of six duodenum samples (Duo6–Duo11) to the T. colubriformis group is notable. These six samples correspond to the sampling conducted in the year 2022, reflecting two-year collection 2020 and 2022 (Supplementary 1). Subsequent PERMANOVA analysis, employing 999 permutations, demonstrated statistically significant differences (P = 0.01, R2 = 0.11) between the groups, confirming the grouping seen in the PCA plot. The level of taxonomy dissimilarity was inferred using Aitchison distances. When comparing the community dissimilarity between paired samples (duodenum-T. colubriformis) and between T. colubriformis against the remaining distances, mean Aitchison distances were determined to be 48.02 and 50.2, respectively (Supplementary Fig. S1).

Principal component analysis (PCA) based on Aitchison distance after centered-log ratio (CLR) transformation.
Figure 4 presents the results of the differential abundance analysis for identifying significant taxa between groups. The multivariable test was calculated using raw ASV count data at the phylum and genus level, resulting in 14 significantly abundant taxa (Supplementary 3). Notably, Mycoplasmoides and Stenotrophomonas, exhibited higher abundance in the helminth when compared directly with duodenal content. Conversely, Succiniclasticum, Saccharofermentans, Lentihominibacter, Butyrivibrio, Ruminococcus, Eubacterium, Pseudobutyrivibrio, Flexilinea, Selenomonas, Anaerotignum, Syntrophococcus, and Blautia were found abundant in the duodenum content. These results show the considerable dominance of the bacteria communities present in the duodenum compared with the microbial communities hosted by T. colubriformis.

The top 10 taxa with differential abundance identified using Multivariable Association with Linear Models (MaAsLin2) test following centered-log ratio (CLP) transformation. Result were consider significant at a false discovery rate (FDR) threshold of < 0.05.
Whole genome sequencing from T. colubriformis bacteria
Two distinct taxa hosted by T. colubriformis were successfully cultured, isolated, and genome sequenced. Comparing these strains with reference genomes in the NCBI database, we identified them as Aeromonas caviae strain 17Tri_A (ANI score of 97.94%) and Aeromonas hydrophila strain 17Tri_Ana3 (ANI score of 96.93%). Both samples showed genomic relatedness exceeding the cutoff range (94–96%). These strains were exclusive to parasites collected from a single sheep. Remarkably, the 16S rRNA V3–V4 analysis further confirmed the absence of Aeromonas sp. in the duodenum of the sheep from where the strains were isolated, documented its exclusive presence within T. colubriformis. The raw data from 16S rRNA V3–V4 sequencing revealed the presence of Aeromonas sp. in seven parasite samples, with a total of 307 ASVs ranging from 1 to 221, with an average of 28 ASVs. Additionally, Aeromonas sp. was identified in the duodenum of four samples, totaling 49 ASVs and ranging from 1 to 33, with an average of 4.5 ASVs.
A. caviae strain 17Tri_A grew under aerobic conditions, the genome size encompassing 4.5 Mb across 109 contigs, containing 4236 genes and 4090 protein-coding. The closest species identified in the database corresponded to the reference genome of A. caviae WP8-S18-ESBL-04 (Ref seq GCF_014169735.1) encompassing 4.6 Mb, containing 4252 genes, of which 4037 are protein-coding. The second bacterium isolated from T. colubriformis was identified as A. hydrophila strain 17Tri_Ana3, demonstrating anaerobic growth capabilities. The genomic content spans 4.8 Mb, encompassing 4502 genes, with 4352 protein-coding sequences distributed among 232 contigs. The taxonomic affiliation of this strain aligns most closely with the Aeromonas hydrophila OnP3.1 (Ref seq GCF_017310215.1) reference genome, covering 4.8 Mb, presenting 4375 genes, with 4207 of them encoding proteins.
We performed an extensive resistome analysis using QMI-AR database to identify antibiotic resistance genes in strains isolated from T. colubriformis. Subsequently, we compared these genes with their respective nearest reference genomes. This database establishes a robust comprehensive framework to annotate and elucidate resistome prediction. Notably, A. caviae 17Tri_A shows a discernibly lower count of antimicrobial resistance (AMR) genes compared with the reference genome (Fig. 5A). Contrary, A. hydrophila 17Tri_Ana3 presented two extras antimicrobial resistance genes including CphA beta-lactamase and MCR phosphoethanolamine transferase (Fig. 5B).

Antimicrobial resistance abundance profile using QIAGEN Microbial Insight-Antimicrobial Resistance database (QMI-AR) (A) Aeromonas caviae (B) Aeromonas hydrophila.
Discussion
Parasitic helminth, including those dwelling in the intestine, often engage in a complex interplay with their host organism. Isolating bacteria from the parasite's intestine can provide insights into the microbial community within the helminth and its potential impact on the host’s health and the parasite’s survival16. In this study, our main goal was to conduct a comparative analysis of microbial diversity and composition between T. colubriformis and the duodenal content from sheep. Additionally, we investigated the presence of fastidious organisms in the harvested T. colubriformis. The samples were obtained from eleven randomly selected sheep, leading to the identification of two distinct strains: Aeromonas caviae strain 17Tri_A and Aeromonas hydrophila strain 17Tri_Ana3. Among the various metrics used to assess genome relatedness such as, DNA-DNA hybridization17, ANI differentiates as a highly reliable indicator of genomic similarity between strains, particularly applied to the identification of these bacteria. Aeromonas spp. are Gram-negative organisms, rod-shaped, facultatively anaerobic, classified within the Gammaproteobacteria class18. Until now, 36 distinct Aeromonas species from different environmental niches have been described, most of which do not exhibit a significant correlation with disease19. Nevertheless, among these species, Aeromonas caviae and Aeromonas hydrophila are recognised as pathogens affecting humans, as they have been isolated from a wide range of intestinal and extra-intestinal infections18. Specifically, A. hydrophila can infect a wide range of fish species causing economic losses to the industry20. While the function of these bacteria in T. colubriformis remains unknown, it is hypothesized to contribute to a symbiotic relationship wherein both the helminth and the bacteria mutually benefit16. Although mutualism relationship is common in nature, there is limited information regarding the specific interactions in the context of parasites and bacteria. The genome sequences from Aeromonas caviae strain 17Tri_A and Aeromonas hydrophila strain 17Tri_Ana3 were further analysed using the QMI-AR database to identify AMR. Interestingly, the analysis revealed a notably reduced numbers of antimicrobial encoding genes in the Aeromonas caviae strain 17Tri_A in comparison to the available reference genomes. This observation may be attributed to the distinct ecological niche of the isolated strains, as the prevalence of AMR genes is known to fluctuate in response to the diverse selective pressure inherent to specific environments21. The prevalence of AMR genes in both stains identified, could be used for control strategies. Another study for example concluded the identification of two highly conserved proteins in Aeromonas species suggesting their potential selection as candidate antigens for developing of vaccines22.
The microbial diversity and composition in T. colubriformis and duodenum content were also investigated in this study. Infection of lambs with gastrointestinal nematodes (Haemonchus contortus and T. colubriformis) changes the microbiome structure. This alteration led to a reduction in the abundance of butyrate-producing microorganisms in ruminal content, specially affecting taxa such as Flavobacteria, Cytophagia, Sphingobacteria, and Negativicutes23. Alpha diversity values showed significant differences in terms of species richness between the duodenum and T. colubriformis. To further explore the dissimilarities between T. colubriformis and duodenum content, the PCA plot based on Aitchison distance matrices revealed two distinct clusters, suggest that some unique ecological factors contributing to differences in terms of species compositions. It is evident that certain duodenum samples exhibit similarities in beta diversity with T. colubriformis. Comparable findings have been observed in the microbiomes of Ascaris suum, demonstrating similarities with the microbiome at the site of infection, particularity at the jejunum and duodenum section24. These results have been also found in Caenorhabditis elegans when contrasted with their corresponding environments25. Specifically, Mycoplasmoides and Stenotrophomonas were found abundant in T. colubriformis, and we could identify them as part of the parasite's microbiota. These genera could contribute to the parasite's ability to colonize and thrive within the host, without underestimating the bacteriological contribution of other taxa present in the parasite. Previous studies have documented the presence of specific bacteria in the gut of different gastrointestinal nematodes such as Trichuris muris, Haemonchus contortus, Teladorsagia circumcincta, Caenorhabditis elegans, and Ascaris suum1,24,25,26,27. For example, in the exploration of the core microbiome of C. elegans, were identified significant abundant members of the family Xanthomonadaceae represented by Stenotrophomonas25. Additionally, the presence of the Xanthomonadaceae family was also observed in the infected larvae L3 stage of Haemonchus contortus and Teladorsagia circumcincta27.
Recent research of intestinal microbial communities in wild Atlantic salmon (Salmo salar) has revealed a general dominance of a single unique Mycoplasma species associated with the biosynthesis of essential amino acids (lysine and threonine) along with thiamine28. Similarly, investigations of the bacterial biodiversity in Anisakis pegreffi, a fish helminth isolated from dolphins (Stenella coeruleoalba), showed that the dominant taxon was the genera Mycoplasma29. While Stenotrophomonas spp. are commonly found widely in the environment, their primary ecological niches are in soil and plants30. Stenotrophomonas spp. possess numerous characteristics with potential for diverse applications in biotechnology. Certain Stenotrophomonas spp. can synthesize antimicrobial compounds for plant protection and produce growth-promoting factors31. Moreover, many Stenotrophomonas spp. exhibit substantial innate resistance to heavy metals and antibiotics, along with a demonstrated ability to degrade various substances, including pollutants32,33,34,35,36. The association between Mycoplasma, Stenotrophomonas spp. and T. colubriformis may involve a range of potential benefits for the nematode, including protection from other microorganisms, enhanced growth, and resistance to environmental factors.
The findings highlight the presence of a distinct bacterial community within T. colubriformis, potentially influencing its persistence in the host's digestive system. The comparison between the duodenal microbiome and the parasite's identified taxa revealed a notable dominance of the gut environment, likely influenced by factors such as feeding behavior, host immune responses, and host-specific attributes. Moreover, this study provides crucial insights into the presence of fastidious organisms in sheep helminth, leading to the isolation and identification of two distinct bacteria, A. caviae and A. hydrophila. Genomic analyses reveal variations in genome size and AMR gene profiles, suggesting potential differences in pathogenicity and responses to antibiotic treatment. Further research elucidating the functional roles and interactions of these identified genera with T. colubriformis is crucial, potentially supporting the way for innovative strategies in controlling parasitic infections.
Materials and methods
Sample collection and processing
In Western Australia, we collected adult T. colubriformis worms from 11 random lambs naturally infected at an abattoir (33° 40′ 24″ S, 117° 31′ 35″ E) when being processed as part of the standard operation during 2020 and 2022. The complete slaughter process is regulated under Australian standard ‘AS: 4696:2007 hygienic production and transport of meat and meat products for human consumption’. Following the slaughter, we promptly tied off and removed the duodenum section from individual sheep on the processing line. The samples were transported immediately to the parasitology laboratory at the Department of Primary Industries and Regional Development (DPIRD), Perth.
At the DPIRD laboratory, the duodenum was dissected, and the total luminal content from each animal was used to collect the parasites. Representative and homogenised gut samples were extracted and stored at -80 °C for subsequent bacterial identification through 16S rRNA analysis. The intestinal contents were passed through a laboratory test sieve (38 Mic, 90 Mic) and the trapped worms were thoroughly washed with tap water (Fig. 6).

Experimental design, including the collection of duodenal content, DNA extraction, 16S rRNA analyses, microbial culture, and bacterial isolation from T. Colubriformis.
Parasite identification and surface cleaning
Helminth were species-identified individually based on the size and morphology of the spicules of male specimens (Fig. 7) in accordance with the Manual of Veterinary Parasitology Laboratory Techniques37. Assigning the female worms to the same species as the males was undertaken, given the inherent difficulty in confidently discerning at the taxonomic level. Specifically, adult female exhibited lengths within the range of 8.6–9.7 mm, while the males, approximately 20% shorter, measured between 6.2 and 6.9 mm38. From each sheep, 400 adult male and female helminth were pooled per animal. Previous experiences indicated that sterilization methods had detrimental effects on the parasite taxa. Therefore, to ensure surface cleaning, we employed sterile phosphate-buffered saline followed by vortex mixer for 1 min. This procedure was repeated several times, with each repetition involving the discarding of the supernatant.

The caudal region of T. colubriformis collected from the duodenal content. The typical male spicules of the adult nematode are shown.
Extraction of DNA from T. colubriformis and duodenal content
T. colubriformis and duodenum content were homogenized separately using 300 helminths and 200 mg of duodenal content per sample in 200 µL cold Tris–EDTA buffer containing 200 mg proteinase K, 1% (v/v) β-mercaptoethanol, 0.5 M EDTA, 10 mg RNAase, and 10% (v/v) SDS, and then incubated at 65 °C for 2 h. An equivalent fraction of phenol:chloroform:isoamyl (25:24:1) solution was added and thoroughly mixed for 1 min and centrifuged at 10,000×g for 5 min. The liquid phase was recovered and relocated into a new tube in an equivalent fraction of chloroform:isoamyl alcohol solution (24:1). The liquid phase was again recovered and transferred into a tube containing ice-cold 95% (v/v) ethanol. The resulting DNA pellet was washed with 70% (v/v) ethanol and resuspended in 50 µL of DEPC water. The quality and quantity of extracted genomic DNA were measured using the NanoDrop 2000 spectrophotometer (Thermofisher, USA) and the Qubit 2.0 fluorometer (Thermofisher, USA). This was followed by electrophoresis on 1% w/v agarose gel to examine genomic DNA integrity.
Microbial culture T. colubriformis
From each of the 11 sheep, we used 100 adult helminths for bacteria culture of fastidious aerobic and anaerobic species. After thorough surface cleaning, performed in previous steps, the parasites were homogenized and plated on Columbia blood agar base (CM0331). The plates were immediately incubated at 37 °C in a laboratory oven to create an aerobic atmosphere. For anaerobic conditions, atmosphere bags (OxoidTM AnaeroGenTM 2.5 L) were placed inside a culture jar at 37 °C. After 24 h, the plates were examined for bacterial growth, and subsequently, two colonies from one sample were isolated based on shape, color, and size. These colonies were then transferred to new sterile agar plates and cultivated for an additional 24 h to obtain pure cultures. DNA from the pure cultures was extracted, purified using the DNeasy® Blood and Tissue Kit (Qiagen Inc.), and stored at − 20 °C for subsequent genome sequencing.
Bacterial genome sequencing and data analysis
Two bacteria colonies were then sequenced on Illumina MiSeq platform. The genomic libraries were prepared using the Nextera XT kit (Illumina, San Diego, USA) following the manufacturer's protocol. The sequencing was performed using a 2 × 300 paired-end protocol. The raw reads' Illumina adapters were initially trimmed using Cutadapt v3.539 assembled with Spades v3.15.540. The annotation was produced by the NCBI Prokaryotic Genome Annotation Pipeline (https://www.ncbi.nlm.nih.gov/genome/annotation_prok/). To identify the most likely species, we employed the Average Nucleotide Identity (ANI) score through the FastANI algorithm v1.3341 by comparing it with the reference genomes presented in NCBI database. The strains were identified based on an ANI range of 94–96% for the species cutoff. Resistome analysis of the two bacterial genomes was conducted using QIAGEN Microbial Insight-Antimicrobial Resistance database (QMI-AR) with ShortBRED algorithm implemented in CLC Genomic Workbench 23.0.5. QMI-AR is compose by ARG-ANNOT, NCBI, CARD, and ResFinder databes.
16S rRNA V3-V4 amplicon library preparation, sequencing, and data analysis
The V3-V4 hypervariable region of the 16S rRNA gene was amplified with the S-D-Bact-0341-b-S-17 (5′-CCTACGGGNGGCWGCAG-3′) and S-D-Bact-0785-a-A-21 (5′-GACTACHVGGGTATCTAATCC-3′) primer pair42. Illumina adapter overhang sequences, 5′-TCGTCGGCAGCGTCAGATGTGTATAAGAGACAG-3′ and 5′-GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAG-3′, were added to the 5′ ends of the forward and reverse primers, respectively. Initial amplification was performed using 30 ng of input DNA, 2 U of MyTaq™ DNA Polymerase (Bioline, Cambridge, UK), 5 × of MyTaq™ reaction buffer, and 10 µM each of the primers under the following conditions: initial denaturation at 95 °C for 1 min, followed by 30 cycles consisting of denaturation at 95 °C, annealing at 55 °C and extension at 72 °C, each for 15 s and then a final extension at 72 °C for 5 min. The PCR products were purified using AMPure XP beads (Beckman Coulter, United States). Indexing PCR was performed with Nextera® XT Index kit (Illumina, United States) using the following protocol: initial denaturation at 95 °C for 3 min, followed by 8 cycles (denaturation at 95 °C, annealing at 55 °C, extension at 72 °C, each for 30 s) and a final extension at 72 °C for 5 min. Sequencing was carried out using the 2 × 300 bp paired-end protocol on an Illumina MiSeq instrument.
Illumina adapters from the raw reads were first trimmed using BBDuk v38.35. In brief, sequence data were then processed with DADA2 v1.22.0 implemented in R43. Filtered and trimmed reads were processed using truncation lengths R1 = 240 bp and R2 = 200 bp after using error rate learning. Merged pairs reads were performed allowing an overlap of 11 bp following chimera removal. Additional filtering based on Amplicon Sequence Variants (ASVs) included: minimum-length = 353, maximum-length = 477, and minimum-count = 10. The ASVs were then taxonomically classified based on the 16S rRNA NCBI database. A minimum confidence score of 80 was used to accept taxonomic assignments at each level. Alpha diversity measurements including ACE, Shannon, Chao1, and Observed OTUs index, were calculated using ampvis2 v2.7.35 after rarefied to a minimum of read depth of 4901. Differences in alpha diversity between the environments were tested using Student’s T-test and Person correlation analysis. For beta assessment, data was transformed using centered log-ratio (CLR) transformation. Beta diversity analysis was tested between the samples using permutational multivariate ANOVA (PERMANOVA) with the vegan v2.6-4. A principal component analysis (PCA) plot based on Aitchison distance matrices was created to visualize the data. Differential associations between the environments were identified using MaAsLin244 v1.15.1 implemented in R, including the following model; min_prevalence = 0.15, max_significance = 0.05, normalization = "CLR", transform = "NONE", reference = c("Environment", "Duodenum"), fixed_effects = c("Environment")).
Data availability
16S rRNA sequence data generated during this study are available in the NCBI SRA accession number PRJNA674764. The genome sequences of Aeromonas caviae strain 17Tri_A and Aeromonas hydrophila strain 17Tri_Ana3 have been deposited in the NCBI under the accession number JAWUZJ000000000 and JAXIOP000000000, respectively.
References
Sinnathamby, G. et al. The bacterial community associated with the sheep gastrointestinal nematode parasite Haemonchus contortus. PLoS ONE 13, 1–25 (2018).
Cortés, A., Rooney, J., Bartley, D. J., Nisbet, A. J. & Cantacessi, C. Helminths, hosts, and their microbiota: New avenues for managing gastrointestinal helminthiases in ruminants. Expert Rev. Anti. Infect. Ther. 18, 977–985 (2020).
Ahmed, N. et al. Molecular and phylogenetic characterization of zoonotic Trichostrongylus species from goats for the first time in Bangladesh. Trans. R. Soc. Trop. Med. Hyg. https://doi.org/10.1093/trstmh/trad034 (2023).
Bhat, A. H., Tak, H., Malik, I. M., Ganai, B. A. & Zehbi, N. Trichostrongylosis: A zoonotic disease of small ruminants. J. Helminthol. 97, e26 (2023).
Du, B. et al. A sheepherder with a severe diarrhea caused by Trichostrongylus colubriformis. Travel Med. Infect. Dis. 48, 102325 (2022).
Cardia, D. F. F., Rocha-Oliveira, R. A., Tsunemi, M. H. & Amarante, A. F. T. Immune response and performance of growing Santa Ines lambs to artificial Trichostrongylus colubriformis infections. Vet. Parasitol. 182, 248–258 (2011).
Bompadre, T. F. V. et al. Trichostrongylus colubriformis infection damages intestine brush board cells and could negatively impact postabsorptive parameters of Santa Ines lambs. Exp. Parasitol. 246, 108464 (2023).
Fernandes, M. A. et al. Computed tomography and radioactive 32P detected phosphorus impairment in metabolism, reduced bones density and animal performance caused by mixed infection of Haemonchus contortus and Trichostrongylus colubriformis in sheep. Vet. Parasitol. 315, 109887 (2023).
Tafere, A., Terefe, G., Mamo, G., Kaba, T. & Shiferaw, J. A comparative study on pathological changes in the small intestine of sheep and goat experimentally infected with Trichostrongylus colubriformis. Vet. Med. Res. Rep. 13, 213–233 (2022).
Dias e Silva, T. P. et al. Trichostrongylus colubriformis infection: Impact on digesta passage rate and lamb performance. Vet. Parasitol. 272, 17–22 (2019).
Dias-Silva, T. P. et al. Computed tomography revealed that bone density in lambs was affected by Trichostrongylus colubriformis infection. Small Rumin. Res. 227, 107044 (2023).
Corrêa, P. S. et al. The effect of Haemonchus contortus and Trichostrongylus colubriforms infection on the ruminal microbiome of lambs. Exp. Parasitol. 231, 108175 (2021).
Cantacessi, C. et al. First transcriptomic analysis of the economically important parasitic nematode, Trichostrongylus colubriformis, using a next-generation sequencing approach. Infect. Genet. Evol. 10, 1199–1207 (2010).
Kraimi, N. et al. Influence of the microbiota-gut-brain axis on behavior and welfare in farm animals: A review. Physiol. Behav. 210, 112658 (2019).
Paz, E. A. et al. Bacterial communities in the gastrointestinal tract segments of helminth-resistant and helminth-susceptible sheep. Anim. Microbiome 4, 23 (2022).
Jenkins, T. P., Brindley, P. J., Gasser, R. B. & Cantacessi, C. Helminth microbiomes—A hidden treasure trove?. Trends Parasitol. 35, 13–22 (2019).
Kim, M., Oh, H. S., Park, S. C. & Chun, J. Towards a taxonomic coherence between average nucleotide identity and 16S rRNA gene sequence similarity for species demarcation of prokaryotes. Int. J. Syst. Evol. Microbiol. 64, 346–351 (2014).
Janda, J. M. & Abbott, S. L. The genus Aeromonas: Taxonomy, pathogenicity, and infection. Clin. Microbiol. Rev. 23, 35–73 (2010).
Fernández-Bravo, A. & Figueras, M. J. An update on the genus Aeromonas: Taxonomy, epidemiology, and pathogenicity. Microorganisms 8, 129 (2020).
Jiravanichpaisal, P., Roos, S., Liu, H. & Söderhäll, K. A highly virulent pathogen, Aeromonas hydrophila, from the freshwater crayfish Pacifastacus leniusculus. J. Invertebr. Pathol. https://doi.org/10.1016/j.jip.2009.02.002 (2009).
Papp, M. & Solymosi, N. Review and comparison of antimicrobial resistance gene databases. Antibiotics 11, 339 (2022).
Zhang, T. et al. The screening of the protective antigens of Aeromonas hydrophila using the reverse vaccinology approach: potential candidates for subunit vaccine development. Vaccines 11, 1266 (2023).
Corrêa, P. S. et al. Tannin supplementation modulates the composition and function of ruminal microbiome in lambs infected with gastrointestinal nematodes. FEMS Microbiol. Ecol. 96, fiaa024 (2020).
Midha, A. et al. Guts within guts: The microbiome of the intestinal helminth parasite Ascaris suum is derived but distinct from its host. Microbiome 10, 229 (2022).
Berg, M. et al. Assembly of the Caenorhabditis elegans gut microbiota from diverse soil microbial environments. ISME J. 10, 1998–2009 (2016).
White, E. C. et al. Manipulation of host and parasite microbiotas: Survival strategies during chronic nematode infection. Sci. Adv. 4, 1–11 (2018).
Hogan, G. et al. Microbiome analysis as a platform R&D tool for parasitic nematode disease management. ISME J. 13, 2664–2680 (2019).
Rasmussen, J. A. et al. Co-diversification of an intestinal Mycoplasma and its salmonid host. ISME J. 17, 682–692 (2023).
Mladineo, I. et al. Microbiota and gut ultrastructure of Anisakis pegreffii isolated from stranded cetaceans in the Adriatic Sea. Parasit. Vectors 12, 1–15 (2019).
Ryan, R. P. et al. The versatility and adaptation of bacteria from the genus Stenotrophomonas. Nat. Rev. Microbiol. 7, 514–525 (2009).
Berg, G., Egamberdieva, D., Lugtenberg, B. & Hagemann, M. Symbiotic plant-microbe interactions: Stress protection, plant growth promotion, and biocontrol by Stenotrophomonas. Life Extrem. Habitats Astrobiol. 17, 445–460 (2010).
Binks, P. R., Nicklin, S. & Bruce, N. C. Degradation of hexahydro-1,3,5-trinitro-1,3,5-triazine (RDX) by Stenotrophomonas maltophilia PB1. Appl. Environ. Microbiol. 61, 1318–1322 (1995).
Zhang, J. F., Zheng, Y. G., Liu, Z. Q. & Shen, Y. C. Preparation of 3-ketovalidoxylamine A C–N lyase substrate: N-p-nitrophenyl-3-ketovalidamine by Stenotrophomonas maltrophilia CCTCC M 204024. Appl. Microbiol. Biotechnol. 73, 1275–1281 (2007).
Juhasz, A. L., Stanley, G. A. & Britz, M. L. Microbial degradation and detoxification of high molecular weight polycyclic aromatic hydrocarbons by Stenotrophomonas maltophilia strain VUN 10,003. Lett. Appl. Microbiol. 30, 396–401 (2000).
Dungan, R. S., Yates, S. R. & Frankenberger, W. T. Transformations of selenate and selenite by Stenotrophomonas maltophilia isolated from a seleniferous agricultural drainage pond sediment. Environ. Microbiol. 5, 287–295 (2003).
Lee, E. Y., Jun, Y. S., Cho, K. S. & Ryu, H. W. Degradation characteristics of toluene, benzene, ethylbenzene, and xylene by Stenotrophomonas maltophilia T3-c. J. Air Waste Manag. Assoc. 52, 400–406 (2002).
Friedhoff, K. T. Manual of Veterinary Parasitological Laboratory Techniques. Veterinary Parasitology Vol. 4 (H.M. Stationery Office, 1978).
Bartley, D. J., Devin, L., Nath, M. & Morrison, A. A. Selection and characterisation of monepantel resistance in Teladorsagia circumcincta isolates. Int. J. Parasitol. Drugs Drug Resist. 5, 69–76 (2015).
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. Journal 17, 10 (2011).
Prjibelski, A., Antipov, D., Meleshko, D., Lapidus, A. & Korobeynikov, A. Using SPAdes de novo assembler. Curr. Protoc. Bioinform. https://doi.org/10.1002/cpbi.102 (2020).
Jain, C., Rodriguez-R, L. M., Phillippy, A. M., Konstantinidis, K. T. & Aluru, S. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 9, 1–8 (2018).
Klindworth, A. et al. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucl. Acids Res. 41, e1–e1 (2013).
Callahan, B. J. et al. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583 (2016).
Mallick, H. et al. Multivariable association discovery in population-scale meta-omics studies. PLoS Comput. Biol. 17, e1009442 (2021).
Acknowledgements
The authors would like to thank the support of WAMMCO Katanning abattoir.
Author information
Authors and Affiliations
Contributions
E.A.P. collected the samples, analyzed the data, and wrote the original draft. E.A.P. and C.Y.T. designed the final experiment. E.G.C. bioinformatic support. D.G.P., J.C.G., S.L., C.CH, S.U.H, G.B.M., and C.Y.T. analyzed the results and corrected the final manuscript. Each author contributed significantly to the manuscript, offering critical input throughout the drafting process, and granted final approval for its publication.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Paz, E.A., Chua, E.G., Palmer, D.G. et al. Revealing the associated microflora hosted by the globally significant parasite Trichostrongylus colubriformis. Sci Rep 14, 3723 (2024). https://doi.org/10.1038/s41598-024-53772-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-53772-z
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
