
Abstract
A metagenomic approach was used to study the gut microbiome of Haemonchus contortus field strains and that of its predilection site, the abomasum of Dohne Merino sheep. The abomasum contents and H. contortus were collected from 10 naturally infected Dohne Merino sheep. The H. contortus specimens were classified and sexually differentiated using morphometric characters and was further confirmed through molecular identification. We investigated differences and similarities between the bacterial composition of the adult male and female H. contortus gut microbiomes, which were both dominated by bacteria from the Escherichia, Shigella, Vibrio and Halomonas genera. Major abundance variations were identified between the shared adult male and female H. contortus microbiomes. The results also revealed that Succiniclasticum, Rikenellaceae RC9 gut group and Candidatus Saccharimonas were the predominant genera in the Dohne Merino abomasum. This study provides insight into the highly diverse bacterial composition of the H. contortus gut microbiome and the Dohne Merino abomasum which needs to be studied further to explore the complex interactions of different gastrointestinal nematode microbiomes with the host.
Similar content being viewed by others


Introduction
Microbiomes are a complex community of microorganisms that vary in their occupied niches, species composition and their influence on various host environments1. These microorganisms live in a close symbiotic relationship with eukaryotic host organisms including nematodes, insects, plants, animals and humans2. The symbiotic relationship between microbes and their hosts range from commensalism, mutualism, parasitism and fatal pathogenic infections3,4. The most extensively researched symbionts of nematodes are those of parasitic plant nematodes and filarial worms, mostly due to their agricultural, biological and medical significance4,5. The most researched symbionts of small ruminants are those of domestic sheep and goats as well as other nematode-bacterial and small ruminant-bacterial symbioses of agricultural interest6,7.
The microbiome occurring within an animal’s gastrointestinal tract (GIT) plays an important role in digestion, nutrition, metabolism and immunity of their host2. Gastrointestinal nematodes (GIN) have a distinct microbiome from their host, and the study of the symbiotic relationship between this microbiome and the GIN host has grown rapidly in recent years, with previous studies reporting on the microbiome associated with Trichuris muris8, Ascaris suum9 and H. contortus4,10. Most of these studies conclusively demonstrate that GIN, in particular the H. contortus harbour microbiome associated with the nematode’s egg-, larval- and adult worm stages performing various functions during their life cycle stages4,10.
The GIN parasite-microbiome symbioses have a measurable impact on the utilization of nutritional resources, maintenance of homeostasis in the digestive tract, and host health10,11. Previous studies on GIN parasite-microbiome interactions have predominantly focused on establishing the microbiome of the host GIT, and the characterization of the microbiome functional roles in metabolism, nutrition and immunity as well as the impact of GIN parasites on host microbiome12. Currently, there is a lack of scientific information on the microbiome harbored by GIN parasites of livestock and their relationship to the host microbiome.
The control of GIN parasites is primarily based on the administration of anthelmintic drugs. The extensive use of anthelmintic drugs leads to the development of drug resistance in GIN parasites, especially the barber’s pole worm H. contortus, which is a highly pathogenic and probably the most economically important nematode infecting small ruminants4,10. Hence, alternative control measures are needed to effectively control GIN parasites. A novel approach based on biocontrol using symbiotic microbial communities has been suggested to limit the use of chemical based treatments13. As such, it is important to understand the dynamics of GIN infections and their respective microbiome in comparison to that of the host which will contribute valuable information towards exploiting the microbiome in an attempt to control GIN in small ruminants4,10,14. The current study focused on the characterization of the adult male and female H. contortus microbiome and that of their predilection site, the abomasum of Dohne Merino sheep.
Results
Morphometrics and molecular analysis of H. contortus isolated from Dohne Merino sheep
A total of 266 GIN, 169 females (63.5%) and 97 males (36.5%) were recovered from 7 out of 10 (70%) abomasum of the Dohne Merino sheep sampled (Table S1 and Fig. S1). A total of 7 pooled adult male H. contortus and 7 pooled female H. contortus samples identified by their morphometric characteristics were confirmed by PCR assay to be H. contortus with an amplification band present at 260 bp (Fig. S3). Care was taken to restrict microbiome contamination (i.e. intact bacteria and their DNA products) originated from the abomasal microbiome (Fig. S2).
Alpha diversity indices of the adult male and female H. contortus and abomasum microbiome
As shown in Table 1, the adult male H. contortus had a higher Operational Taxonomic Unit (OTU) richness than the female (p-value < 0.05). However, when compared to the abomasum, the abomasum microbiome had the highest richness (p-value < 0.05). There was a clear difference between the adult male H. contortus gut microbiome in comparison to the female gut microbiome across all alpha diversity indices except for Faith’s Phylogenetic Diversity (PD) which did not show any significant difference (p-value > 0.05) (Table 1 and Fig. S4). Similarly, in Fig. 1A, the abomasum content was compared to the adult male H. contortus and the female microbiome, and displayed a significant difference in Observed OTUs, Shannon and Simpson indices (p-value < 0.05).

Principal coordinate analysis (PCoA) based on unweighted UniFrac distances (B). Alpha diversity (A) matrices, observed OTUs, Shannon, Simpson of the abomasum content, adult male and female H. contortus microbiome at 97% identity. Data represent average values from pooled populations.
Beta diversity indices of the adult male and female H. contortus and abomasum microbiome
The Principal Coordinates Analyses (PCoA) results indicated that adult male and female H. contortus microbiome clustered together. The abomasum microbiome clustered on its own and was separated from the adult male and female H. contortus microbiome cluster (Fig. 1B). Furthermore, the unweighted (Fig. 1B) and weighted UniFrac (Fig. S5) distance analysis indicated that the microbiome composition of the abomasum was significantly different from that of the adult male and female H. contortus (p-value < 0.05) (Table S3). The microbiome composition was also significantly different between the abomasum content and the adult female H. contortus (p-value < 0.05). The abomasum and adult male H. contortus microbiome was also found to be significantly different (p-value < 0.05) (Table S3).
Microbiome composition at the phylum level
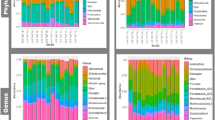
A total of 23 different phyla were identified in the adult male H. contortus microbiome which were predominantly dominated by bacterial phyla: Proteobacteria (57%), Firmicutes (24%), Bacteroidetes (8%) and Actinobacteria (6%) (Fig. 2A). In the adult female H. contortus microbiome, a total of 17 different phyla were identified with the dominant phyla being Proteobacteria (94%) followed by Firmicutes (3%), Bacteroidetes (1%), Actinobacteria (0.6%) and Cyanobacteria (0.3%) (Fig. 2B). Analysis of the abomasum identified a total of 20 different phyla. The five most abundant bacterial phyla in the abomasum were Firmicutes (45%) followed by Bacteroidetes (26%), Actinobacteria (7%), Patescibacteria (4%) and Planctomycetes (3%) (Fig. 2C).

Krona plot for taxonomic abundance of the adult male (A), female (B) H. contortus and Abomasum (C) microbiome at the phylum level. Data represent average values from pooled populations.
Microbiome composition at the genus level
The adult male H. contortus microbiome was dominated by Vibrio (15%) followed by Escherichia-Shigella (8%), Halomonas (8%) and Acinetobacter (3%) (Fig. 3A). The predominant genera in the adult female H. contortus microbiome was Escherichia-Shigella (28%), Vibrio (11%), Halomonas (6%), Acinetobacter (2%) and Alteromonadales (2%) (Fig. 3B). The abomasum was dominated by Succiniclasticum (5%), Rikenellaceae RC9 gut group (4%), Candidatus Saccharimonas (4%), Ruminococcaceae UCG-011 3%, p-1088-a5 gut group (3%) and Prevotella 1 (3%) (Fig. 3C).

Krona plot for taxonomic abundance of the adult male (A), female (B) H. contortus and Abomasum (C) microbiome at the genus level. Data represent average values from pooled populations.
Unique and shared microbiome composition
In total, 660 unique bacterial OTUs across the three hosts were identified (Table S4). The abomasum content had higher bacterial OTU abundance followed by the adult female H. contortus whilst the male had the lowest bacterial OTU abundance (Fig. S6). The results showed a total of 57 bacterial OTUs shared between the adult male and female H. contortus. The adult male H. contortus and abomasum shared 113 OTUs with each host having 98 and 169 unique OTUs present, respectively. A total of 16 bacterial OTUs were shared between adult female H. contortus and the abomasum (Fig. 4A).

Venn diagram (A) and krona plot depicting the composition and number of unique and shared OTUs associated with adult male (B), female (C) H. contortus and abomasum content (D) microbiome. Data represent average values from pooled populations.
Unique genera in the microbiome
The unique genera in the adult male H. contortus were found to be Corynebacterium 1 (10%), Azospirilum (7%), Lysinibacillus (4%), Lactococcus (4%), Acetobacter (4%), Empedobacter (3%) and Paeniclostridium (3%) (Fig. 4B). The adult female H. contortus contained the following unique genera: Lactobacillus (15%), Bradyrhizobium (7%), Pseudonocardia (5%), Prevotella (5%), Anaplasma (4%) and Treponema2 (4%) (Fig. 4C). Unique genera in the abomasum were CPla-4 termite group (22%), Pirellula (4%), Lachnospiraceae AC2044 group (3%), Prevotellaceae UGG003 (3%), Prevotellaceae UGG004 (3%), Prevotella (3%) and Coprostanoligenes group (3%) (Fig. 4D).
Discussion
The barber’s pole worm (H. contortus) is a common parasitic nematode infecting small ruminants and one of the most pathogenic GIN of sheep15,16,17. Identification of common stomach worms, including H. contortus, in small ruminants has been previously based on recognized morphological characteristics that make it possible to differentiate between Haemonchus species, particularly, H. contortus and H. placei. The spicule lengths and vulvar flap provide the quickest and easiest characteristics to be used for separating populations of H. contortus from H. placei.18,19,20. In the present study, the recovered Haemonchus spp. specimens were identified as adult male and female H. contortus based on their morphological characteristics as suggested by Lichtenfels et al.21 and Gibbons18. The adult female H. contortus were recognised mainly by their reddish colour and vulvar flap features. The adult male H. contortus were identified based on the low variability of spicule morphology within species. The morphological classification of the specimens was confirmed by PCR assay using species specific H. contortus primers 22.
Following recovery of the parasites from the abomasum, a rigorous treatment consisting of antibiotic solution (1 mg/ml ampicillin/1 mg/ml gentamicin) and 4% sodium hypochlorite was used to ensure removal of all microbiome contamination on the parasite surface. The PCR assay results with 16S rRNA gene primers further served as an indicator that there was no contamination on the parasite originating from the abomasal microbiome. These results corroborated the previously published results by White23 and El-Ashram and Suo10 who reported that sterilisation with this method is sufficient to remove abomasal microbiome contaminants on the surface of the GIN.
The adult male and female H. contortus microbiome were characterised in the absence of external microbiome contaminants on the surface of the parasite. The results indicated that the adult female H. contortus microbiome was dominated by bacterial phyla: Proteobacteria (94%) followed by Firmicutes (3%), Bacteroidetes (1%), Actinobacteria (0.6%) and Cyanobacteria (0.3%). The adult male H. contortus microbiome was dominated by bacterial phyla: Proteobacteria (57%) followed by Firmicutes (24%), Bacteroidetes (8%), Actinobacteria (6%) and Cyanobacteria (2%). These results are similar to those observed in earlier studies conducted on Trichuris muris by White23, who reported that the adult T. muris microbiome was dominated by bacterial phyla: Bacteroidetes followed by Firmicutes and Proteobacteria. El-Ashram and Suo10 and Sinnathamby et al.4 reported that the H. contortus microbiome was dominated by the bacterial phyla: Proteobacteria followed by Firmicutes, Tenericutes and Actinobacteria, and Firmicutes followed by Proteobacteria and Bacteroidetes respectively. Relatedly, the microbiome composition at phylum level observed in the current study on H. contortus were similar to those observed in previous studies of T. muris, Ascaris suum and H. contortus from United Kingdom, United States of America and China respectively. However, there were variations in abundance of these phyla which are usually influenced by the type of host they inhibit and environmental conditions10,23. White23, El-Ashram and Suo10 and Sinnathamby et al.4 compared the differences and similarity in the microbiome of the different parasite life-cycle stages but not of the different genders of the adult stages and the host. This study therefore provided additional information by comparing the adult male and female H. contortus microbiome as well.
On a genus level, the adult female H. contortus microbiome was dominated by bacterial genera: Escherichia-Shigella (28%) followed by Vibrio (11%), Halomonas (6%), Acinetobacter (2%) and Alteromonadales (2%). The dominant genera in the adult male H. contortus microbiome were Vibrio (15%) followed by Escherichia-Shigella (8%), Halomonas (8%) and Acinetobacter (3%). The predominant bacterial genera in the adult male H. contortus were similar to female but with variation in the genera abundance. The adult female H. contortus showed higher genera abundance than male H. contortus, although the adult male H. contortus had more diversity than female in terms of the overall microbiome. Among the aforementioned genera, Escherichia- Shigella, Vibrio, Halomonas and Acinetobacter were predominantly shared by both adult male and female H. contortus. These results are different from those reported by El-Ashram and Suo10 who indicated that the predominant bacterial genera of the H. contortus were Escherichia-Shigella (87.5%), Pseudomonas (5.91%) and Ochrobactrum (5.06%), which we believe is caused by the type of host they infect and environmental conditions of their locations.
The microbiome of the adult male and female H. contortus showed similarity and variations in terms of abundances and diversity in the present study. These results indicated that H. contortus harbours a distinct microbiome compared to their host. There was significant difference between adult male and female H. contortus microbiome diversity and abundances. The H. contortus samples in this study were recovered from the same sheep breed, fed under the same diet and under similar environmental conditions. The variations in the microbiome of adult male H. contortus and female may be caused by hormonal changes, immune response or the reproductive needs, including egg development in females, which does not occur in male GINs. The presence of the microbiome in H. contortus suggests a potential nutritional importance of the microbiome to the worms, either providing metabolites that the worm cannot make itself, or aiding the breakdown of ingested blood meal.
The Dohne Merino abomasum microbiome naturally infected with H. Contortus field strains were characterised to gain insight into the impact that GIN parasites infection has on the host microbiome and how these interactions influence H. contortus microbiome. The abomasum microbiome was characterised and found to be dominated by the following bacterial phyla: Firmicutes (45%), Bacteroidetes (26%), Actinobacteria (7%), Patescibacteria (4%) and Planctomycetes (3%). Similar observations were reported in previous studies on small ruminants which indicated that the abomasum was dominated by Bacteroidetes, Firmicutes, Actinobacteria and Proteobacteria.24,25. With regard to the abundance of these predominant phyla, there were variations found across the studies. Interestingly, the phyla Patescibacteria and Planctomycetes were found in abundance in the abomasum content in the present study with none of these phyla reported in other previously published studies23,24,26. These variations between the present study and previously published studies could be due to known factors that affect microbiome composition such as parasite burden, breed type, diets and different environmental condition.
The predominant assigned bacterial genera observed in the abomasum content were Succiniclasticum (5%), Rikenellaceae RC9 gut group (4%), Candidatus Saccharimonas (4%), Ruminococcaceae UCG-011 3%, p-1088-a5 gut group (3%) and Prevotella 1 (3%). These predominant genera are known to specialize in complex polysaccharides degradation, such as starch and cellulose, and contribute in the metabolism of dietary fiber in ruminants27. These results agree with observations reported by Zeng et al.25, Tanca et al.28 and Zhang et al.27 whereby predominant genera in the GIT depend on the parasite burden, diet and environmental condition. Hence, there were variations in the microbiome observed in the present study and previously published studies25,27,28.
The results further indicated that all abomasum content microbiome clustered together, suggesting that the sheep abomasum harbour similar microbiome. The adult male and female H. contortus microbiome clustered together, indicating that they harbour similar microbiome as well, which could be due to similar living environmental conditions and diet. El-Ashram and Suo10 reported similar results which shows the H. contortus eggs and adult stage microbiome cluster together because they share similar environmental conditions inside the host. When compared, the adult male H. contortus and female appeared to have lower bacterial diversity than the abomasum content. Hence, the H. contortus microbiome does not simply reflect their host abomasum content microbiome, although they spend majority of their life-cycle in the host abomasum. A previous study by Sinnathamby et al.4 reported that the different parasite life-cycle stages microbiomes are different from that of their predilection site. Moreover, El- Ashram and Suo10,Sinnathamby et al.4 also reported that during the H. contortus life cycle development stages, the egg stage, L1, and L2 and L3 interact with different microbiome from their respective environment, thus each stage might have distinct microbiome that they pick up from their environment and the host.
Furthermore, we found that the adult male H. contortus harbour a higher number of unique bacterial OTUs compared to the female and shared a higher number of bacterial OTUs with the abomasum content than the female. These results suggest that there are various selective measures at play within the H. contortus digestive tract, different to that of the host gastrointestinal tract. Therefore, some bacterial species are unable to persist and others thrive. The metabolic needs of the H. contortus might vary to that of its host and microbiome selection will depend on what GIN parasite require for its own fitness and survival within the host. These results correspond with Sinnathamby et al.4 observations whereby they reported that the H. contortus harbour distinct/unique microbiome from its host, which are important for their survival. Furthermore, the present study indicated that the abomasum content harbour higher OTUs abundance compared to H. contortus, which would be due to its higher metabolic needs. This observation is similar to previous studies on T. muris and H. contortus by White et al.29 and Sinnathamby et al.4 which reported that the host harbour more diverse microbiome than the parasite.
Previous studies by Lu et al.30,Gómez-Garzón et al.31 and Abdel-Salam et al.32 reported that bacterial strains, Pseudoalteromonas marina, Vibrio attantica strain 5–16, Pseudomonas aeruginosa and beteli, and Lysinibacillus can produce nematicidal volatile compounds with activities against the root-knot nematode Meloidogyne incognita and pine wood nematodes. Interestingly, similar bacterial genera were observed in the unique OTUs in the present study as well. As result, these bacteria can be manipulated in livestock production system due to their nematicidal activity to target pathogenic GIN in livestock such as H. contortus and potentially become biological control agents for GIN parasites while reducing the use of chemical based treatments31,32. The presence of these bacteria gives hope that it is possible to use symbiotic microbiome to control GIN parasites in livestock.
In conclusion, the adult male H. contortus appears to harbour more diverse microbiome as compared to the female. The abomasum has more diversity compared to both adult male and female H. contortus. Data obtained in this study has shown presence of bacterial genera (Vibrio, Lysinibacillus and Anaplasma) in the H. contortus and abomasum that can produce nematicidal volatile compounds that are active against nematodes, with possibility to be manipulated to control populations of GIN parasites within the sheep abomasum. To determine the exact nature of microbiome-H. contortus and host-H. Contortus microbiome interaction, additional and complementary analyses are necessary to uncover specific molecular mechanisms underlying the nematode-microbiome as a method of controlling nematodes.
Materials and methods
Ethical approval
Ethical approval was obtained from the Onderstepoort Veterinary Institute Animal Ethics Committee (AEC) (AEC No: 36.18) and the scientific committee of the Integrated Pest Management subprogram, North-West University. All methods in this study were approved by AEC as well as the scientific committee of the Integrated Pest Management subprogram, North-West University, and performed in accordance with the relevant guidelines and regulations.
Animals and parasites
The study was conducted using 10 Dohne Merino sheep (Table S1), collected from Wauldby farm (E27° 37′ 32.7"’’ S32° 35′ 07′’) which consist of a flock of over 450 ewes situated in the Stutterheim district in the Eastern Cape Province, South Africa. This farm has a well-documented history of natural H. contortus infection.
Recovery and morphological characterization of gastrointestinal parasites
Naturally infected Dohne Merino sheep were slaughtered under humane conditions. The abomasum was detached from the rest of the GIT and the contents of the abomasum including adult worms were collected4,33. The worms were recovered from 7 of the 10 sampled Dohne Merino sheep. The worms were first examined in detail with the assistance of an experienced helminthologist to identify and sexually differentiate worms based on their morphological characters as described by Lichtenfels et al.20 and Jacquiet et al.34 (Fig. S1). The morphological identification was further confirmed by PCR assay using species specific primers (Table S2).
Sterilisation of parasitic H. contortus surface
The samples were washed with phosphate-buffered saline (PBS), pH 7.4 and incubated in an antibiotic solution (1 mg/ml ampicillin/1 mg/ml gentamicin) for 2 h to kill external bacteria. Thereafter, the worms were washed three times with 4% sodium hypochlorite for 20 s, followed by washing five times with sterile PBS4,10. This process was done to remove external and abomasal contaminates on the surface of the adult male and female H. contortus. A PCR assay was employed to confirm the absence of microbiome contamination on the GIN surfaces (Fig. S2).
Microbial DNA isolation
A total of 24 samples were collected from 10 Dohne Merino sheep. Of the 24 samples, 10 of them were abomasum content samples, 7 were pooled adult male and 7 pooled female H. contortus worms which were then homogenized and incubated in lysis solution composed of 20 mM Tris·HCl, pH 8.0, 2 mM Sodium EDTA, 1.2% Triton X-100, plus 20 mg/ml Lysozyme at 37 °C for 60 min. Microbial DNA together with GIN parasite DNA was extracted from the pre-processed abomasum contents and the sterile H. contortus samples using the QIAamp DNA Microbiome Kit (QIAGEN, Hilden, Germany), as described in the manufacturer’s protocol. The DNA samples were stored at − 20 °C until further processing.
Amplicon library preparation and sequencing
A region of approximately 470 bp to 500 bp encircling the V3-V4 hypervariable region within the 16S rRNA gene was amplified using a set of commonly used primers for the analysis of the gut microbiome (Table S2). The region of 260 bp was also amplified using species specific primers for confirmation of H. contortus (Table S2)22. Briefly, each 25 μl of PCR assay reaction contained 5 μl of genomic DNA as the template, 12.5 μl 2× KAPA HiFi HotStart ReadyMix (Kapa Biosystems, United States) and 5 μl of 10 μM of each primer. PCR reactions were carried out in a BIO-RAD T100TM Thermal Cycler (Bio-Rad Laboratories, United Kingdom) using the following protocols: (1) for microbiome, an initial denaturation step performed at 95 °C for 5 min followed by 30 cycles of denaturation (95 °C, 30 s), annealing (56 °C, 30 s) and extension (72 °C, 40 s), and a final elongation of 10 min at 72 °C; (2) for H. contortus, an initial denaturation step performed at 95 °C for 5 min followed by 30 cycles of denaturation (95 °C, 30 s), annealing (58 °C, 30 s) and extension (72 °C, 40 s), and a final elongation of 10 min at 72 °C. The PCR amplicon were assessed by gel electrophoresis in 1% agarose gel run at 100 V for 45 min, and the sizes of the products were validated by comparison with a molecular marker. The PCR products were cleaned-up using AMPure XP beads (Beckman Coulter, United States). Amplicon library preparation and sequencing were conducted as per Illumina 16S Metagenomic Sequencing Library Preparation guide. The prepared pooled libraries were then loaded into the Illumina MiSeq sequencing platform.
Pre-processing and data analysis
De-multiplexed, paired-end fastq reads and a mapping file or metadata file were imported into the Quantitative Insights Into Microbial Ecology 2 program (QIIME2, ver. 2018.6.0, https://qiime2.org/). Sequencing reads were pre-processed, quality filtered and analysed using the Divisive Amplicon Denoising Algorithm 2 (DADA2) software package, wrapped in QIIME2 version 2018.6.0. Resultant feature sequences were summarized and then annotated using the QIIME2 q2- feature-classifier plugin and Silva 132 99% OTUs (full-length, seven-level taxonomy) classifier pre-trained to the full-length Silva database to assign taxonomy to all ribosomal sequence variants.
Statistical analysis
Alpha and beta-diversity analyses were performed with the q2-diversity plugin wrapped in QIIME2 at a sampling depth of 15,000 sequences. Alpha diversity metrics (Shannon diversity index, Simpson diversity index, Pielou’s measure of species evenness and Faith’s Phylogenetic Diversity) were calculated and statistical significance determined using Kruskal–Wallis tests. The Beta diversity was calculated by Bray–Curtis and Jaccard distances, unweighted and weighted UniFrac metrics. Principal Coordinate analyses (PCoA) was performed based on unweighted and weighted UniFrac distances and visualized with QIIME2 emperor visualizer (ver. 2018.6.0) and the statistical significances were calculated using Permutational Multivariate Analysis Of Variance (PERMANOVA). Additional analysis were performed with in-house python scripts (Python version 3.6. 1, 2017-03-21), KronaTools and RStudio software using phyloseq package (R version 3.5.0, 2018-04-23).
Data availability
The datasets generated during and/or analysed during the current study are available in the NCBI Sequence Read Archive (SRA) repository, Accession number: PRJNA698475.
References
Stubbendieck, R. M., Vargn-Bautista, C. & Straight, P. D. Bacterial communities’ interactions to scale. Front. Microbiol. 7, 1234 (2016).
Kreisinger, J., Bastien, G., Hauffe, H. C., Marchesi, J. & Perkins, S. E. Interactions between multiple helminths and the gut microbiota in wild rodents. Philosophical transactions of the Royal Society of London. Ser. Biol. Sci. 370(1675), 20140295 (2015).
Moya, A., Pereto, J., Gil, R. & Latorre, A. Learning how to live together: genomic insights into prokaryote- animal symbioses. Nat. Rev. Genet. 9, 218–229 (2008).
Sinnathamby, G. et al. The bacterial community associated with the sheep gastrointestinal nematode parasite Haemonchus contortus. PLoS ONE 13(2), e0192164 (2018).
Kanfra, X., Liu, B., Beerhues, L., Sørensen, S. J. & Heuer, H. Free-living nematodes together with associated microbes play an essential role in apple replant disease. Front. Plant Sci. 9, 1666 (2018).
Huang, J., Li, Y. & Lu, Y. Bacterial community in the rumen of Tibetan sheep and Gansu alpine fine-wool sheep grazing on the Qinghai-Tibetan Plateau, China. J. General Appl. Microbiol. 8, 003 (2016).
Wang, J., Fan, H., Han, Y., Zhao, J. & Zhou, Z. Characterization of the microbial communities along the gastrointestinal tract of sheep by 454 pyrosequencing analysis. Asian-Austral. J. Anim. Sci. 30(1), 100–110 (2017).
Hayes, K. S. et al. Exploitation of the intestinal microflora by the parasitic nematode Trichuris muris. Science 328, 1391–1394 (2010).
Shahkolahi, A. M. & Donahue, M. J. Bacterial flora. A possible source of serotonin in intestines of adult female Ascaris suum. J. Parasitol. 79, 17–22 (1993).
El-Ashram, S. & Suo, X. Exploring the microbial community (microflora) associated with ovine Haemonchus contortus (macroflora) field strains. Sci. Rep. 7, 70 (2017).
Fisher, R. M., Henry, L. M., Cornwallis, C. K., Kiers, E. T. & West, S. A. The evolution of host-symbiont dependence. Nat. Commun. 8, 15973 (2017).
Glendinning, L., Nausch, N., Free, A., Taylor, D. W. & Mutapi, F. The microbiota and helminths: sharing the same niche in the human host. Parasitology 141, 1255–1271 (2014).
Sahoo, A. & Khan, F. A. Nutritional and biological control synergism against gastrointestinal nematodes in small ruminants. J. Vet. Sci. Anim. Husb. 4(1), 104 (2016).
Fukruksa, C. et al. Isolation and identification of Xenorhabdus and Photorhabdus bacteria associated with entomopathogenic nematodes and their larvicidal activity against Aedes aegypti. Parasites Vectors 10, 440 (2017).
Regassa, F., Teshale, S., Reta, D. & Yosef, K. Epidemiology of gastrointestinal parasites of ruminants in Western Oromia. Int. J. Appl. Res. Vet. Med. 4(1), 51–57 (2006).
Roeber, F., Jex, A. R. & Gasser, R. B. Impact of gastrointestinal parasitic nematodes of sheep, and the role of advanced molecular tools for exploring epidemiology and drug resistance an Australian perspective. Parasites Vectors 6, 153 (2013).
Tsotetsi, A. M. & Mbati, P. A. Parasitic helminths of veterinary importance in cattle, sheep and goats on communal farms in the North Eastern Free State, South Africa. J. S. Afr. Vet. Assoc. 74(2), 45–48 (2003).
Gibbons, L. M. Revision of the genus Haemonchus Cobb 1898 (Nematoda: Trichostrongylidae). Syst. Parasitol. 1, 3–24 (1979).
Lichtenfels, J. R., Pilit, P. A. & Le jambre, L. F. Cuticular ridge patterns of Haemonchus contortus and Haemonchus placei (Nematoda: Trichostrongyloidea). Proc. Helminthol. Soc. Wash. 53(1), 94–101 (1986).
Lichtenfels, J. R., Pilitt, P. A. & Hoberg, E. P. New morphological characters for identifying individual specimens of Haemonchus spp. (Nematoda: Trichostrongyloidea) and a key to species in ruminants of North America. J. Parasitol. 80(1), 107–119 (1994).
Lichtenfels, J. R., Pilit, P. A. & Le jambre, L. F. Spicule lengths of the ruminant stomach nematodes Haemonchus contortus, Haemonchus placei, and their hybrids. Proc. Helminthol. Soc. Wash. 55(1), 97–100 (1988).
Amarante, M. R. V., Santos, M. C., Bassetto, C. C. & Amarante, A. F. T. PCR primers for straightforward differentiation of Haemonchus contortus, Haemonchus placei and their hybrids. J. Helminthol. 91(6), 757–761 (2017).
White, E. C. Infection by the gastrointestinal parasite Trichuris muris: defining the microbiota of the parasite and the host. Preprint at https://www.research.manchester.ac.uk/portal/files/54589495/FULL_TEXT.PDF (2016).
Wang, J., Fan, H., Han, Y., Zhao, J. & Zhou, Z. Characterization of the microbial communities along the gastrointestinal tract of sheep by 454 pyrosequencing analysis. Asian-Austral. J. Anim. Sci. 30(1), 100–110 (2016).
Zeng, Y. et al. Microbial community compositions in the gastrointestinal tract of Chinese Mongolian sheep using Illumina MiSeq sequencing revealed high microbial diversity. AMB Exp. 7(1), 75 (2017).
Zeng, Y. et al. Characterization of the cellulolytic bacteria communities along the gastrointestinal tract of Chinese Mongolian sheep by using PCR-DGGE and real- time PCR analysis. J. Microbiol. Biotechnol. 31, 1103–1113 (2015).
Zhang, F. et al. Caenorhabditis elegans as a model for microbiome research. Front. Microbiol. 8, 485 (2018).
Tanca, A. et al. Diversity and functions of the sheep faecal microbiota: a multiomic characterization. Microb. Biotechnol. 10(3), 541–554 (2017).
White, E. C. et al. Manipulation of host and parasite microbiotas: survival strategies during chronic nematode infection. Sci. Adv. 4(3), eaap7399 (2018).
Lu, H. et al. Identification and nematicidal activity of bacteria isolated from cow dung. Ann. Microbiol. 64(1), 407–411 (2014).
Gómez-Garzón, C., Hernández-Santana, A. & Dussán, J. A genome-scale metabolic reconstruction of Lysinibacillus sphaericus unveils unexploited biotechnological potentials. PLoS ONE 12(6), e0179666 (2017).
Abdel-Salam, M. S., Amee, H. H., Soliman, G. M., Elkelany, U. S. & Asar, A. M. Improving the nematicidal potential of Bacillus amyloliquefaciens and Lysinibacillus sphaericus against the root-knot nematode Meloidogyne incognita using protoplast fusion technique. Egypt. J. Biol. Pest Control 28, 31 (2018).
Simpson, H. V., Simpson, B. H., Simcock, D. C., Reynolds, G. W. & Pomroy, W. E. Abomasal secretion in sheep receiving adult Ostertagia circumcincta that are prevented from contact with the mucosa. N. Z. Vet. J. 47, 20–24 (1999).
Jacquiet, P., Cabaret, J., Cheikh, D. & Thiam, E. Identification of Haemonchus species in domestic ruminants based on morphometrics of spicules. Parasitol. Res. 83(1), 82–86 (1996).
Acknowledgements
We thank Waudby farm for supplying us with the Dohne Merino sheep. The National Research Council (NRF) is hereby acknowledged for funds made possible towards this project, NRF-Thuthuka funding, Grant Number 113988.
Author information
Authors and Affiliations
Contributions
Conceptualization, O.M.M.T., A.M.T., P.S., C.A.H; supervision; O.M.M.T., A.M.T., P.S., R.E.P; writing original draft preparation, T.M.; methodology; T.M. manuscript review and editing, O.M.M.T., A.M.T., P.S., C.A.H., F.C.M., R.E.P.; funding acquisition, P.S; bioinformatics and statistical analysis, T.M., R.E.P. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mafuna, T., Soma, P., Tsotetsi-Khambule, A.M. et al. Bacterial profiling of Haemonchus contortus gut microbiome infecting Dohne Merino sheep in South Africa. Sci Rep 11, 5905 (2021). https://doi.org/10.1038/s41598-021-85282-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-85282-7
This article is cited by
-
A diverse microbial community and common core microbiota associated with the gonad of female Parascaris spp.
Parasitology Research (2024)
-
A detailed workflow to develop QIIME2-formatted reference databases for taxonomic analysis of DNA metabarcoding data
BMC Genomic Data (2022)
-
Bacterial communities in the gastrointestinal tract segments of helminth-resistant and helminth-susceptible sheep
Animal Microbiome (2022)
-
The microbial community associated with Parascaris spp. infecting juvenile horses
Parasites & Vectors (2022)
-
Microbial community in resistant and susceptible Churra sheep infected by Teladorsagia circumcincta
Scientific Reports (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
