
Abstract
Notch signaling determines cell fates in mouse intestine. Notch receptors contain multiple epidermal growth factor-like (EGF) repeats modified by O-glycans that regulate Notch signaling. Conditional deletion of protein O-fucosyltransferase 1 (Pofut1) substantially reduces Notch signaling and markedly perturbs lineage development in mouse intestine. However, mice with inactivated Pofut1 are viable, whereas complete elimination of Notch signaling in intestine is lethal. Here we investigate whether residual Notch signaling enabled by EGF-domain-specific O-linked N-acetylglucosamine transferase (Eogt) permits mice conditionally lacking Pofut1 in intestine to survive. Mice globally lacking Eogt alone were grossly unaffected in intestinal development. In contrast, mice lacking both Eogt and Pofut1 died at ~ 28 days after birth with greater loss of body weight, a greater increase in the number of goblet and Paneth cells, and greater downregulation of the Notch target gene Hes1, compared to Pofut1 deletion alone. These data reveal that both O-fucose and O-GlcNAc glycans are fundamental to Notch signaling in the intestine and provide new insights into roles for O-glycans in regulating Notch ligand binding. Finally, EOGT and O-GlcNAc glycans provide residual Notch signaling and support viability in mice lacking Pofut1 in the intestine.
Similar content being viewed by others


Introduction
The small intestine of male and female mammals is comprised of differentiated epithelial lineages derived from Lgr5hi-expressing and potentially other intestinal stem cells (ISC), which maintain overall mucosal homeostasis and provide principal functions in nutrient uptake and metabolism1,2. Notch signaling controls the functioning of ISCs by regulating maintenance of the stem cell pool, and specifying lineage differentiation to absorptive enterocyte cells or secretory goblet, Paneth, enteroendocrine and Tuft cells3. The critical role of Notch signaling is mechanistically complex, however, dependent on post-translational modifications of Notch receptors and ligands that fine tune the strength of Notch signals known to be central in embryonic and tissue development4. Fundamental to this fine tuning are epidermal growth factor-like (EGF) repeats present in the extracellular domain of Notch receptors (NECD) that are modified by O-glycans including O-fucose and O-GlcNAc glycans5,6 (Fig. 1A).

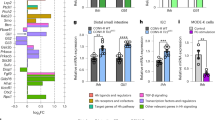
O-glycans on NOTCH receptors and expression of Eogt and Pofut1 in mouse intestine. (A) Diagram of mouse NOTCH1 and NOTCH2 ECDs showing O-fucose and O-GlcNAc glycans predicted at EGF repeats that contain the appropriate amino acid consensus sequence: C2XXXXS/TC3 for O-fucose and C5XX(G/S/P)(Y/F/T)T/SGXXC6 for O-GlcNAc glycans (indicated by the respective initiating sugar). O-glucose glycans at a third consensus site are not shown. NRR Notch regulatory region. (B) Eogt and Pofut1 expression in mouse intestinal cells by scRNAseq. The trajectory analysis of the combined data from intestinal cells of 3 mice is shown. The definition of each intestinal cell type is given in Abbreviations. (C) Transcript levels of candidate genes expressed in each cell type were quantitated for 3 mice.
These O-glycans regulate Notch signaling in cell-based assays, in mouse models and in humans4,7,8,9. Only ~ 50 proteins contain one or more EGF repeats with appropriate consensus sites for O-glycan addition10, and not all O-glycans necessarily affect biological function11. However, O-fucose on EGF12 of NOTCH1 interacts directly with Notch ligands DLL412 and JAG113 to facilitate Notch-ligand interactions and the loss of this fucose is embryonic lethal in the C56BL6/J background14. Importantly, amongst proteins with modifiable EGF repeats, Notch receptors contain by far the greatest number, and thus phenotypes arising from the loss of O-glycan glycosyltransferases commonly reflect defective Notch signaling4,7,8,9.
In the intestine, conditional deletion (cKO) by Villin-Cre of Pofut1, the gene encoding the transferase that adds O-fucose to appropriate EGF repeats, causes a substantial reduction in Notch signaling and a marked increase in secretory cell lineages in male and female mice15. Pofut1 cKO mice are smaller than littermates but survive for at least 6 months. Mice lacking a single Fringe gene (Lfng or Rfng) encoding transferases that add GlcNAc to the O-fucose transferred to EGF repeats by POFUT1, also exhibit altered Notch signaling and a Notch-defective intestinal phenotype16. A clear illustration of the importance of modulating Notch signaling, rather than Notch acting as an “on–off switch”, is that mice with down-regulation of Notch by cKO of Pofut1 survive for ≥ 6 months on a mixed genetic background15, whereas cKO of two Notch ligand genes Dll1 and Dll4 targeted by Villin-Cre causes death at 4–6 days after the final tamoxifen dose17. Similarly, conditional deletion of Notch1 and Notch218, or treatment with a gamma-secretase inhibitor19, produce severe inhibition of Notch signaling in the intestine. Interestingly, no apparent effects of Jag1 deletion in intestinal epithelium were observed17.
Given the viability of Pofut1[F/F]:Villin-Cre mice, we hypothesized that residual Notch signaling may be enabled by O-GlcNAc glycans on Notch receptors. EOGT is the EGF domain specific GlcNAc transferase that initiates modification of EGF repeats by O-GlcNAc glycans20. Eogt null pups exhibit altered development of the postnatal retina typical of defective Notch signaling, and EOGT regulates Notch receptor-ligand interactions21. Therefore, a dual approach was used to investigate interactive roles of O-fucose and O-GlcNAc glycans on Notch receptors. First, Chinese hamster ovary (CHO) cells lacking Eogt, Pofut1 or both Eogt and Pofut1 were generated by a CRISPR/Cas9 strategy and examined for binding of canonical Notch ligands. Second, small intestine of male and female mice in which Eogt and Pofut1 were genetically inactivated individually or in combination were interrogated. The data reveal that, while mice lacking Eogt from conception did not exhibit an obvious intestinal phenotype, marked Notch signaling defects in Pofut1 cKO intestine were significantly enhanced when both Eogt and Pofut1 were inactivated. The combined data provide evidence for synergy between O-fucose and O-GlcNAc glycans in regulating Notch signaling in the intestine.
Results
Eogt, Pofut1, Notch receptors and ligands expressed in mouse intestine
Notch signaling is fundamental in regulating the balance of cell interactions necessary for normal development. Therefore, fine tuning of Notch signaling is essential and is achieved by post-translational modification of Notch receptors by glycosylation7,8,9. Figure 1A illustrates consensus sites of predicted glycosylation of mouse NOTCH1 and NOTCH2 by O-fucose initiated by POFUT1, and by O-GlcNAc initiated by EOGT5,6,22,23, and the subsequent extension by different enzymes. O-glucose glycans that also decorate Notch receptor EGF repeats are not shown. O-fucose and O-GlcNAc glycans are recognized by Notch ligands whereas O-glucose glycans probably are not24. Roles for Notch signaling have been clearly documented in intestinal stem cell functions17,18, but fine tuning in the intestine has not been defined. Single cell RNAseq (scRNAseq) of mouse intestinal epithelium identified complex relationships among expression of Notch1 and Notch2 receptor genes, Delta1 and Delta4 Notch ligand genes and Eogt and Pofut1 glycosyltransferase genes (Fig. 1B,C). While Notch1 and Notch2 were expressed mainly in stem and progenitor cells, the genes encoding Dll1 and Dll4 were predominantly expressed in early goblet cells, with DLL4 also expressed in early enterocytes. Importantly, Pofut1 and Eogt were expressed in the same cell types as Notch receptors and ligands, and also more heterogeneously. We therefore investigated further roles for Notch signaling in intestinal homeostasis by genetic inactivation of Eogt and/or Pofut1 to determine differential impacts on the differentiation of cells along the crypt-villus axis. But first we investigated effects of O-glycan removal on Notch ligand binding in a cell-based assay using CHO cells.
Notch ligand binding to CHO O-glycan mutants
To investigate potential synergism between O-fucose and O-GlcNAc glycans (Fig. 1A), deletion mutants were generated by a CRISPR/Cas9 strategy in Lec1 CHO cells25. Lec1 CHO cells do not make hybrid or complex N-glycans and therefore provide a simplified glycosylation environment for glycosyltransferase gene deletions affecting O-glycan synthesis26. Lec1 cells harboring inactivated Eogt, Pofut1 or Eogt and Pofut1 genes were characterized as described in Methods and Supplementary Figure S1. To determine if the cell surface expression of NOTCH1 (as an example of Notch receptors) was affected by the loss of O-glycan subsets from its extracellular domain (NECD1), Lec1 and CRISPR/Cas9 Lec1 mutants were assessed for binding of an anti-NECD1 antibody by flow cytometry. Modestly reduced expression of cell surface NOTCH1 was observed in Eogt:Pofut1 dKO Lec1 cells (Fig. 2A,B). In contrast, the binding of soluble Notch ligands DLL1-Fc and DLL4-Fc was markedly reduced in both Pofut1 and Eogt:Pofut1 mutant Lec1 cells (Fig. 2A,B). The reduction in DLL4-Fc binding to Eogt:Pofut1 dKO compared to Pofut1 cKO cells had a P value of 0.056 in the unpaired, one-tailed Student’s t test (Fig. 2B). Although not meeting a significance of P < 0.05, the difference suggested potential synergism between O-fucose and O-GlcNAc glycans. This hypothesis was pursued in vivo.

Notch ligand binding to Lec1 cells lacking EOGT, POFUT1 or both. (A) Representative flow cytometry profiles for anti-NOTCH1 ECD and Notch ligands DLL1-Fc and DLL4-Fc binding to Control (gray), Eogt KO, Pofut1 KO (blue) and Eogt:Pofut1 dKO (red) Lec1 cells. Secondary antibody alone for Pofut1 KO (black line) and Eogt:Pofut1 dKO (gray line). (B) Mean fluorescence index (MFI) for binding of anti-NOTCH1 ECD or different Notch ligands to Eogt KO, Pofut1 KO or Eogt:Pofut1 dKO CHO cells normalized to MFI of Control cells (n = 3–4 experiments). P values from one-way ANOVA followed by Tukey’s multiple comparisons test—*P < 0.05, ***P < 0.001, ****P < 0.0001. For DLL4-Fc binding, the unpaired, one-tailed Student’s t test P value for Pofut1 cKO versus Eogt:Pofut1 dKO was 0.056.
Eogt supports residual Notch signaling in Pofut1 cKO intestine
Intestinal development was investigated in mice homozygous for Eogt inactivation compared to heterozygotes and wild type littermates. There were no differences in body weight, length of small intestine, production of secretory cells, or morphology and dimensions of villi or crypts in Eogt[+/+], Eogt[+/−] and Eogt[−/−] mice (Supplementary Figure S2). To investigate potential alterations at the molecular level, intestinal crypts were prepared by fractionation as described in Methods. The relative enrichment of Lyz1 in crypts (fraction IV) and Fabp2 in villi (fraction I) was determined by qRT-PCR (Supplementary Figure S3A,B). Notch1 and Lgr5 transcripts were reduced by homozygous inactivation of Eogt (Supplementary Figure S3C–G), indicating an impact of the loss of Eogt on Lgr5+ stem cells which express Notch1 (Fig. 1B). However, this did not lead to altered expression of Notch signaling target genes such as Hes1 in Eogt null crypts. Finally, in all morphological, histological and molecular parameters examined, wild type and Eogt[+/−] intestine were indistinguishable, and thus Eogt heterozygous mice were appropriate controls for evaluating effects in mice with compound mutations in both Eogt and Pofut1.
Conditional inactivation of Pofut1 in the mouse intestine greatly inhibits canonical Notch signaling, but the mice are viable and live to ≥ 6 months of age, albeit with significantly altered lineage representation15. To test the hypothesis that EOGT and O-GlcNAc glycans provide residual Notch signaling in Pofut1 cKO mice, we compared intestinal development in Pofut1 cKO versus Eogt:Pofut1 dKO mice. Eogt[−/−]Pofut1[F/F] and Eogt[+/−]Pofut1[F/+]:Villin-Cre mice were crossed to produce compound genotypes (Supplemental Figure S4). The progeny obtained and those surviving to P28 after birth are shown in Table 1. All potential genotypes were detected at P8 and reflected the expected Mendelian inheritance ratios. However, by P28 all Eogt:Pofut1 dKO pups had died (Chi-squared significance < 0.025). This indicated that Eogt contributed to development of intestinal epithelium in Pofut1 cKO pups.
To investigate effects earlier in postnatal development, histopathology of small intestine was assessed at P15. There were no changes in body weight, villi length or crypt depth in small intestine of Eogt KO, Pofut1 cKO or Eogt:Pofut1 dKO pups compared to control mice at P15 (Supplementary Figure S5A–C). The numbers of Alcian Blue-positive goblet cells and eosin-stained Paneth cells in the small intestine of Eogt KO mice were similar compared to controls (Supplementary Figure S5D–G). In contrast, goblet cells significantly increased in crypts and villi of Pofut1 cKO and Eogt:Pofut1 dKO pups (Supplementary Figure S5D–F). Importantly, the number of goblet cells per crypt was significantly higher in Eogt:Pofut1 dKO compared to Pofut1 cKO intestine, indicating a contribution of Eogt to maintaining secretory cell development. Consistent with this, there was also an increase in Paneth cells in Eogt:Pofut1 dKO crypts compared to Pofut1 cKO crypts (Supplementary Figure S5G). Thus, while deletion of Eogt had no apparent effects at P15, cKO of Pofut1 caused a shift to greater secretory cell differentiation which was exacerbated by the concomitant elimination of Eogt in Eogt:Pofut1 dKO pups.
Since Eogt:Pofut1 dKO mice died about 1 month after birth, intestinal development was investigated at P28. Body weight and length of small intestine were significantly decreased in Eogt:Pofut1 dKO compared to Pofut1 cKO mice (Fig. 3A,B). Both goblet and Paneth cells were significantly increased in Eogt:Pofut1 dKO compared to Pofut1 cKO intestine (Fig. 3C–F). Villus length was significantly decreased in Eogt:Pofut1 dKO intestine compared to control, Eogt KO and Pofut1 cKO intestine (Supplementary Figure S6A). Crypt depth, crypt length and crypt width were increased in Pofut1 cKO and Eogt:Pofut1 dKO compared to control or Eogt KO intestine (Supplementary Figure S6B,D). Thus, for all parameters assessed, conditional deletion of Pofut1 from mice lacking Eogt in intestine caused a more severe phenotype than deletion of Pofut1 alone, indicating an important effect of Eogt in complementing canonical Notch signaling.

Synergy between Pofut1 and Eogt in intestinal homeostasis. (A,B) Body weight and length of small intestine (SI) of Control, Eogt KO, Pofut1 cKO and Eogt:Pofut1 dKO mice (n ≥ 12 mice per group). (C,D) Number of goblet cells and area of Paneth cells in the crypts of experimental mice (~ 50 crypts were analyzed in 4 mice per group). (E,F) Representative images showing goblet and Paneth cells in the small intestine of experimental mice (n = 4 mice per group). P values from one-way ANOVA followed by Tukey’s multiple comparisons test—*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. Scale bar 50 μm.
Notch activation is markedly reduced in Pofut1 cKO and Eogt:Pofut1 dKO crypts
Notch signaling is active at the base of crypts within Notch receptor-expressing ISC supported by ligand-expressing Paneth cells16. Cleaved NICD1 (activated NOTCH1) is mainly detected in ISC and transit amplifying (TA) cells located in the crypt base27, and our scRNAseq trajectory analysis of Notch receptor and ligand expression are consistent with this (Fig. 1B,C). To investigate Notch signaling status in crypt cells, NOTCH1 cleavage to NICD1 was determined by immunohistochemistry (Fig. 4A) and western blot analysis (Fig. 4B). Crypts of Pofut1 cKO and Eogt:Pofut1 dKO intestine were enlarged due to a significant increase in secretory cells and a concomitant reduction in crypt base columnar ISC. Val1744 antibody detects cleaved NOTCH1 and positive cells were rare to non-existent in both Pofut1 cKO and Eogt:Pofut1 dKO crypts (Fig. 4A). Control sections showed abundant signal for cleaved NOTCH1 in nuclei. Specificity of signals for antibody to NICD1 was confirmed in sections stained with secondary antibody alone (Supplementary Figure S7). Further, western blot analyses showed activated NOTCH1 and NOTCH2 in control crypt cells, but essentially no signal for NICD1 or NICD2 in either Pofut1 cKO or Eogt:Pofut1 dKO crypt lysates (Fig. 4B, Supplementary Figure S8) with no significant differences detectable between the single and double mutant samples.

Expression of activated, cleaved NOTCH receptors in intestine. (A) Immunohistochemistry of intestinal sections for binding of antibody to cleaved NOTCH1 (NICD1) in crypts of Control, Eogt KO, Pofut1 cKO and Eogt:Pofut1 dKO mice (n = 1–3 mice per group). (B) Western blots for cleaved NICD1 and NICD2 in the crypts of Control, Pofut1 cKO and Eogt:Pofut1 dKO (representative images of 4–6 mice per group). Scale bar 20 μm.
Notch ligand binding to Eogt KO, Pofut1 cKO and Eogt:Pofut1 dKO ISC
To investigate Notch ligand binding to Notch receptors in ISC, crypts were isolated, single cell suspensions prepared, and analyzed by flow cytometry. Viable, single cells were gated on CD44+CD45−CD24− ISC (Supplementary Figure S9). Cell surface expression of NOTCH1 was determined using antibody to NECD1, and binding of soluble Notch ligands carrying a C-terminal Fc tag was determined using anti-Fc antibody. Three experiments were performed, each including ISC from control, Eogt KO, Pofut1 cKO and Eogt:Pofut1 dKO mice. Representative flow cytometry profiles and histograms of mean fluorescence index (MFI) are shown in Fig. 5. MFI data for mutant ISC were normalized to control ISC in each experiment. The expression of NOTCH1 on ISC cell populations from mutant mice was variable and did not differ significantly among groups (Fig. 5D). Binding of DLL1-Fc but not DLL4-Fc was significantly reduced compared to control in Pofut1 cKO and Eogt:Pofut1 dKO ISC. Furthermore, DLL1-Fc binding to Eogt:Pofut1 dKO ISC was significantly lower than to Pofut1 cKO ISC.

Notch ligand binding to intestinal stem cells. (A–C) Flow cytometry profiles of binding of anti-NOTCH1 ECD, DLL1-Fc and DLL4-Fc to Eogt KO, Pofut1 KO or Eogt:Pofut1 dKO intestinal stem cells. (D) MFI for anti-NOTCH1 ECD, Notch ligand DLL1-Fc and Notch ligand DLL4-Fc (n = 3 mice) binding to Control, Eogt KO, Pofut1 KO and Eogt:Pofut1 dKO normalized to MFI of Control cells. Average MFI for NOTCH1 ECD binding was Control, 3639; Eogt KO, 6245; Pofut1 KO, 4623; Eogt:Pofut1 dKO, 5837. Average MFI for DLL1-Fc binding was Control, 213,525; Eogt KO, 184,167; Pofut1 KO, 143,871; Eogt:Pofut1 dKO, 105,452. Average MFI for DLL4-Fc binding was Control, 39,905, Eogt KO, 29,691; Pofut1 KO, 20,562; Eogt:Pofut1 dKO, 40,779. Blue profiles show NOTCH1 ECD or Notch ligand-Fc binding, gray profiles show secondary antibody binding. P values from one-way ANOVA followed by Tukey’s multiple comparisons test—*P < 0.05, ***P < 0.001, ****P < 0.0001.
Notch target and Notch pathway gene expression in Eogt:Pofut1 dKO intestine
To investigate the functional impact of EOGT and POFUT1 synergism, expression of Notch signaling target genes, lineage and ISC marker genes and Notch pathway member genes were investigated in crypt cells. Fractionation produced a similar enrichment of crypt versus villi cells regardless of genotype (Supplementary Figure S4A,B). Hes1 is directly regulated by Notch signaling and is decreased by reduced Notch signaling in the intestine, leading to increased expression of targets such as Math115,28. Consistent with this, Hes1 expression was markedly decreased in Pofut1 cKO crypts, and importantly, was significantly further decreased in Eogt:Pofut1 dKO crypts (Fig. 6A). In parallel, Math1 expression increased in both Pofut1 cKO and Eogt:Pofut1 dKO crypts (Fig. 6A). Furthermore, transcripts of secretory cell marker genes ChgA and Lyz1 were increased in Pofut1 cKO and significantly more so in Eogt:Pofut1 dKO crypts (Fig. 6B) as expected from the increased secretory cells in Pofut1 cKO intestine, and the even greater increase in Eogt:Pofut1 dKO intestine (Fig. 3,E). Muc2 transcripts were also increased, more in Eogt:Pofut1 dKO than Pofut1 cKO crypts (Fig. 6B). Expression of two canonical stem cell marker genes, Lgr5 and Olfm4 were both markedly decreased in single and double mutant crypts (Fig. 6C). Transcripts of Notch1 were decreased in both Pofut1 cKO and Eogt:Pofut1 dKO crypts, while Notch2 expression was decreased in only Eogt:Pofut1 dKO crypts (Fig. 6D). Jag1 expression was also decreased in both mutants (Fig. 6D). In contrast, Dll1 expression was increased in Eogt:Pofut1 dKO crypts while Dll4.

Notch pathway genes expressed in crypts of Pofut1 cKO and Eogt:Pofut1 dKO mice. (A) qRT-PCR performed on cDNA from intestinal crypts. Transcript levels of key Notch pathway target genes in Control (white), Pofut1 cKO (light gray) and Eogt:Pofut1 dKO (dark gray) crypt cells. Transcript levels of marker genes expressed in (B) different intestinal cell lineages, or (C) ISC. (D) Transcript levels of Notch receptors and Notch ligands (n = 4 mice per group). P values from one-way ANOVA followed by Tukey’s multiple comparisons test—*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, or unpaired, two-tailed Student’s t-test with Welch’s correction *P < 0.05, **P < 0.01.
expression was increased in both mutants. Jag2 transcripts were also increased in both of the mutants, and increased to a greater extent in Eogt:Pofut1 dKO compared to Pofut1 cKO crypts (Fig. 6D). Overall, therefore, the expression of marker genes of Notch signaling support the hypothesis that EOGT contributes to canonical, Notch ligand-mediated signaling in maintaining homeostasis of the intestinal mucosa.
Discussion
Here we show that Notch signaling in mouse intestine is fine-tuned by the addition of two types of O-glycan to Notch receptors that modulate the binding of canonical Notch ligands—O-fucose and O-GlcNAc glycans. While the contribution to Notch signaling strength of POFUT1 and O-fucose glycans is much stronger than that of EOGT and O-GlcNAc glycans, the necessity for the latter became clear in Eogt:Pofut1 dKO intestine. Whereas Pofut1:Villin1-Cre cKO mice exhibit severely defective Notch signaling in mouse intestine15 (and herein), Pofut1 cKO mice live for ≥ 6 months. By contrast, we establish here that Eogt:Pofut1:Villin1-Cre dKO mice die at ~ 1 month with a more severely Notch signaling defective intestinal phenotype. Nevertheless, these compound mutants live longer than mice lacking both Notch1 and Notch2 or Dll1 and Dll4 which die within days of conditional knockout17,18. We propose that limited Notch signaling persists in Eogt:Pofut1 dKO intestine via Notch receptors that are modified by only O-glucose glycans. While O-glucose glycans do not appear to interact directly with Notch ligands12,13,24, they are important in regulating Notch receptor trafficking to the cell surface29,30. Since anti-NOTCH1 ECD antibodies and soluble Notch ligands DLL1-Fc and DLL4-Fc bound to ISC from Eogt:Pofut1 dKO intestine (Fig. 5), Notch receptors were clearly present at the cell surface. We propose that O-Glucose glycans on Notch receptors support their trafficking in Eogt:Pofut1 dKO intestine, and that elimination of Notch signaling by reduced glycosylation would require the triple knockout of Pofut1, Eogt and Poglut1.
Revealing that Eogt contributes to Notch signaling in mouse intestinal development is important for several reasons. First, to better understand mechanistic bases of regulation of the ubiquitously important Notch signaling pathway. Second, to understand potential pathologies in individuals with the congenital disease of glycosylation EOGT-CDG (Adams-Oliver Syndrome Type 4; AOS4). Those afflicted lack a functional EOGT gene and exhibit a range of limb extremity and scalp developmental defects as well as ocular, vascular and intellectual abnormalities consistent with defective Notch signaling31,32. In the mouse embryo, the Eogt gene is first expressed in limb buds and the apical ectodermal ridge before expression in digit condensates at ~ E12.5, consistent with defective development of fingers and toes in EOGT-CDG patients31. The involvement of Eogt in mouse intestinal development reported here is a potential indication of the possibility of perturbed intestinal development or function in EOGT-CDG patients. Mutations in EOGT have been associated with 2% of 10,967 colorectal adenocarcinoma patients queried33.
Determining how EOGT and the O-GlcNAc glycans fine tune Notch functions in the intestinal environment is challenging. Ideally, NOTCH1 from ISC in the various mouse cohorts would be purified and the nature of the linked O-glycans defined, but this is currently not feasible due to the low concentration of NOTCH1 and NOTCH2 in ISC. The first O-glycan analysis of mouse NOTCH1 from a physiologic source required stimulation of splenic T cells in culture to increase Notch1 expression and obtain sufficient NOTCH1 for mass spectrometric analysis34. Roles for the O-glycans on canonical Notch ligands must also be evaluated. Deletion of Rfng is reported to reduce modification of Delta ligands and their ability to stimulate Notch signaling in ISC16. However, determining which under-glycosylated partner in a defective Notch receptor-ligand interaction is responsible can only be done when the other partner is fully glycosylated. Thus, proving that Notch ligands require specific O-glycans to induce Notch signaling necessitates conditionally deleting the target glycosylation gene in ligand-expressing cells. Finally, to conclude that Notch signaling is affected by the deletion of a glycosyltransferase requires evidence that the loss generates: (1) a phenotype that mimics loss of Notch pathway member(s); (2) causes alterations in Notch ligand binding; and (3) causes altered expression of Notch signaling target genes. Here we provide this combined evidence, demonstrating that EOGT and the O-GlcNAc glycans it initiates support Notch signaling during intestinal development in the absence of POFUT1 and O-fucose glycans.
Methods
Single cell RNAseq bioinformatics
Bioinformatic trajectory analysis of scRNAseq data obtained from Epcam+CD45− C57Bl6/J adult intestinal crypt cells and deposited as GSE188339 was performed as described35.
Primers and antibodies
Primer sequences are given in Supplementary Table 1 and antibodies are given in Supplementary Table 2.
Generation of Chinese hamster ovary (CHO) glycosylation mutants
CHO Lec125 cells with inactivated Pofut1 were previously described as Pofut1 KO-1 (P4–11) and KO-2 (P4–15)36. The Pofut1 deletion was confirmed by western blotting with a rabbit anti-bovine POFUT1 antibody37. To inactivate Eogt, two guide RNAs (GTATGACTACTCCAGCCTCC in exon 1; Eogt2 GTTTGCAGCTATGTCGACGT in exon 2), an ATTO 550-labeled tracrRNA and Cas9 (all from Integrated DNA Technologies, Inc., Coralville, IA) were lipofected into Lec1 cells. After 24 h, ATTO 550+ cells were sorted by flow cytometry and the 3% most positive cells were seeded at 1 cell per well in alpha minimal essential medium (αMEM, Thermo Fisher Scientific, Waltham, MA) containing 10% fetal bovine serum (FBS) and 1% Penicillin–Streptomycin. Lec1 Pofut1 KO-1 cells were transformed by lipofection with the same Eogt gRNAs to generate Eogt:Pofut1 dKO mutants. After 8–10 days, colonies were expanded and characterized. Genomic DNA PCR detected Eogt alleles (Supplementary Figure S1A) and RT-PCR detected cDNA products (Supplementary Figure S1B) as described21. Mutants were designated E116 (Eogt KO) and PE316 (Eogt:Pofut1 dKO).
Notch ligand binding to CHO mutants
Cells (~ 0.5 × 106) were washed with FACS binding buffer (FBB; Hank’s balanced salt solution (MilliporeSigma Corning, Burlington, MA), 2% bovine serum albumin (BSA) fraction V (GeminiBio, West Sacramento, CA), 1 mM CaCl2 and 0.1% sodium azide) and incubated in 50 μl of rat anti-mouse CD16/CD32 Fc blocker (Supplementary Table 2 for all antibodies) in FBB for 15 min on ice. Sheep anti-mouse NOTCH1 (100 μl diluted 1:50 in FBB) or 100 μl FBB containing 0.75 μg DLL1-Fc, DLL4-Fc or JAG1-Fc soluble Notch ligand described previously37 was added to the Fc block and incubated on ice for 1 h, washed twice with cold FBB and incubated with secondary antibody in FBB (1:100 rhodamine Red-X anti-sheep IgG) or anti-Fc for ligands (1:100, Alexa Flour 488 anti-human IgG) for 30 min on ice. Secondary antibody alone determined background binding. Following addition of 5 μl 7-AAD in 95 μl FBB for 10 min at room temperature, FBB (250 μl) was added, cells filtered into 5 ml polystyrene tubes and analyzed by flow cytometry using a FACS Caliber flow cytometer and FlowJo software (BD Biosciences, Franklin Lakes, NJ).
Mouse models
Mice with a targeted inactivating mutation in the Eogt gene were developed at Nagoya University and previously described21. Pofut1[F/F] mice were also generated previously38. Mice expressing Villin-Cre (B6.Cg-Tg(Vil1-cre)1000Gum/J, JAX strain 021504) were from Jackson Laboratories, Portland, MA. Cross breeding generated Pofut1 cKO (Eogt[+/−]:Pofut1[F/F]:Villin-Cre), Pofut1:Eogt dKO (Eogt[−/−]Pofut1[F/F]:Villin-Cre) and mutants lacking Villin-Cre. Genomic DNA PCR was used to genotype. Body weight was recorded following euthanasia, small intestine was dissected, length measured, and tissue processed. Experimental protocols were approved by the Albert Einstein Institutional Animal Care and Use Committee (IACUC) under protocol numbers 20170709 and 00001311. All experiments were performed in accordance with the relevant rules and regulations in the approved experimental protocols and this study is reported in accordance with the ARRIVE guidelines (https://arriveguidelines.org). Mice were euthanized by asphyxiation in a CO2 chamber followed by cervical dislocation.
Isolation of crypt and villus fractions
Small intestine was washed with cold phosphate-buffered saline containing 1 mM CaCl2, 1 mM MgCl2 and 1 mM dithiothreitol (PBS (Ca/Mg/DTT), sectioned into four, flushed extensively with PBS (Ca/Mg/DTT), one end tied, everted with a gavage needle, filled to ~ 75% with PBS (Ca/Mg/DTT) and tied off before washing with buffer A (96 mM NaCl, 1.5 mM KCl, 27 mM Na-Citrate, 8 mM KH2PO4, 5.6 mM Na2HPO4 and 1 mM DTT) at 37 °C with constant shaking for 10 min. Tissue was shaken with buffer B (1.5 mM EDTA, 0.5 mM DTT and 0.1% BSA) four times at 37 °C for 10, 10, 30 and 20 min, respectively. Buffer B collected at each time was centrifuged to give fractions I-IV. Relative enrichment of villi and crypts was determined by qRT-PCR of marker genes (Supplementary Figure S2A,B). Fractions were snap frozen in liquid N2 and stored at − 80 °C. Conditional deletion of Pofut1 by Villin-Cre was validated by PCR genotyping (Supplementary Figure S2C).
Histopathology and immunohistochemistry
Jejunum was fixed in 10% neutral buffered formalin for 48 h and processed through a graded series of alcohol to prepare paraffin blocks. Sections (5 μm) were stained with hematoxylin and eosin or Alcian Blue. Slides were scanned using a 3D Histech P250 High-Capacity Slide Scanner. CaseViewer 2.4 was used to measure villi length, crypt depth, crypt width and crypt length. QuantCenter software was used to quantitate goblet and Paneth cell staining. For immunohistochemistry, 5 μm sections were dipped into xylene and graded concentrations of ethanol as described (http://www.abcam.com/protocols/). Slides were boiled in sodium citrate for 20 min, incubated with 0.1% Triton X-100 followed by 1.5% H2O2 in Tris-buffered saline (TBS) for 20 min. Antibody incubations were performed at room temperature in a humidified chamber as follows: blocking in 10% FBS with 1% BSA in TBS for 1 h, followed by primary antibody overnight in 1% BSA in TBS, washing with TBS containing 0.025% Triton X-100, and incubation in HRP-conjugated secondary antibody diluted in TBS with 1% BSA for 1 h. Diaminobenzidine peroxidase substrate kit (Vector Laboratories, Burlingame, CA) treatment was followed by counter staining with Hematoxylin and Bluing reagent. Slides were treated with a graded ethanol series and xylene and mounted using Permount.
Quantitative RT-PCR (qRT-PCR)
Total RNA was extracted from ~ 107 frozen cells using TRIzol (Thermo Fisher Scientific, Waltham, MA). RNA was dissolved in RNAse-free water and 1 µg estimated using Nanodrop was used to make 20 µl cDNA (Verso cDNA synthesis kit, Thermo Fisher Scientific). cDNA was amplified using the PowerUp SYBR Green master mix (Thermo Fisher Scientific) and 750 nM each primer. Vii7 Real-Time PCR system (Applied Biosystems, Foster City, CA) was used to run qRT-PCR for 40 cycles. Each sample was run in triplicates using 384 wells plate. Gene expression was calculated relative to Gapdh and Hprt by log2ddCT method.
Western blotting
Frozen cells (~ 107) were homogenized in 100 μl lysis buffer containing 1% IGEPAL, 1%TX-100, 0.5% Deoxycholate (all from Sigma-Aldrich, St. Louis, MO), and Roche complete™ Protease Inhibitor (Sigma-Aldrich) and incubated on ice for 30 min. After centrifugation at 5000g for 5 min at room temperature, the supernatant in a new tube received 20% glycerol and protein concentration was estimated by Bradford’s Dye Reagent Concentrate (BioRad Laboratories, Hercules, CA). For gel electrophoresis, 50–100 µg protein was analyzed by SDS-PAGE, transferred to PVDF membrane and blocked in 5% Non-fat dry milk in Tris-buffered saline 0.05% Tween 20 for one hour at room temperature. Membranes were then cut at the mid-point between the 75 and 50 kDa visible molecular weight markers and then were overnight incubated separately with relevant primary antibodies in blocking buffer at 4 °C. Membrane rinsed with Tris-saline was incubated with HRP-secondary antibody in the same buffer for one hour at room temperature. Enhanced chemiluminescent substrate was used and signals visualized on X-ray film (Thermo Fisher Scientific).
Notch ligand binding to ISC
Washed small intestine pieces were scraped to remove villi, transferred to 20 ml ice-cold 2 mM EDTA and 2 mM glutamine in PBS (Ca/Mg free) and incubated on ice for 20 min. Tissue was then transferred to fresh 20 ml PBS (Ca/Mg free) with 2 mM glutamine and shaken by hand for 30 s. This process was repeated another four times. The last four washes were filtered through a 70 μm strainer into 1% BSA/PBS (Ca/Mg free)-coated 50 ml falcon tubes. Filtrates were spun at 1500 rpm for 10 min at 4 °C to pellet crypts. Single cells were prepared in 3 ml enzyme-free dissociation buffer (Gibco, Thermo Fisher Scientific, Waltham, MA) incubated in a 37 °C water bath for 10 min, with pipetting every 30 s, then 7 ml of alpha MEM was added, cells were filtered through a 40 μm strainer, pelleted and resuspended in Zombie NIR dye in PBS (1:7000; Biolegend). After 30 min rocking at 4 °C, cells were washed with PBS, fixed in 4% paraformaldehyde (PFA; Emsdiasum, Hatfield, PA) in PBS (Ca/Mg free) 15 min at 4 °C with rocking, washed 3 times with cold FBB and stored in FBB at 4 °C.
Flow cytometry performed within 1–6 days investigated NOTCH1 expression using anti-NECD1 antibody AF5267 and Notch ligand binding for DLL1-Fc (R&D Systems, Inc., Minneapolis, MN) and DLL4-Fc (Aro Biosystems, Newark, DE). Briefly, ~ 106 fixed cells were resuspended in 40 μl CD16/CD32 Fc blocker in FBB (1:40) and incubated 15 min on ice. Antibodies in FBB to CD45 (1:400), CD44 (1:800), CD24 (1:800), CD166 (1:800) and GRP78 (1:800) with anti-NOTCH1 or 2 μg of Notch ligand-Fc were added to cells in FcR block and incubated 30 min on ice. Cells with no anti-NOTCH1 or ligand-Fc determined background. Cells were washed and incubated with secondary antibody (1:600 rhodamine Red-X conjugated donkey anti-sheep IgG or 1:100 Fc-specific anti-IgG-Dylight 405) for 30 min on ice, washed, resuspended in 300 μl FBB, filtered into flow tubes and 300,000 events were recorded in a CytekTM Aurora flow cytometer, analysis by FlowJo software (BD Biosciences).
Statistical analysis
Data are represented as mean ± SEM. GraphPad Prism 9.0.1 was used to perform one-way ANOVA followed by Tukey’s multiple comparisons tests or unpaired two-tailed Student’s t tests with Welch’s correction as noted. Observed and expected allele inheritance was analyzed at ~ P8 and of survival at ~ P28 by the Chi-squared test.
Data availability
The data presented in this paper will be made available upon request to Mohd Nauman and/or Pamela Stanley.
Abbreviations
- BSA:
-
Bovine serum albumin
- CHO:
-
Chinese hamster ovary
- cKO:
-
Conditional knockout
- DAB:
-
3,3′-Diaminobenzidine
- Div:
-
Dividing
- dKO:
-
Double knockout
- DTT:
-
Dithiothreitol
- EC:
-
Enterocyte
- EDTA:
-
Ethylenediamine tetra-acetic acid
- EE:
-
Enteroendocrine
- EGF:
-
Epidermal growth factor-like
- FBS:
-
Fetal bovine serum
- gRNA or crRNA:
-
Guide RNA
- ISC:
-
Intestinal stem cell
- NECD1:
-
NOTCH1 extracellular domain
- NICD1:
-
NOTCH1 intracellular domain
- R:
-
Replicating
- RNP:
-
Ribonucleoprotein
- sc:
-
Single cell
- TA:
-
Transit amplifying
- UK:
-
Unknown
References
Barker, N. Adult intestinal stem cells: Critical drivers of epithelial homeostasis and regeneration. Nat. Rev. Mol. Cell Biol. 15, 19–33. https://doi.org/10.1038/nrm3721 (2014).
Gehart, H. & Clevers, H. Tales from the crypt: New insights into intestinal stem cells. Nat. Rev. Gastroenterol. Hepatol. 16, 19–34. https://doi.org/10.1038/s41575-018-0081-y (2019).
Liang, S. J., Li, X. G. & Wang, X. Q. Notch signaling in mammalian intestinal stem cells: Determining cell fate and maintaining homeostasis. Curr. Stem Cell Res. Ther. 14, 583–590. https://doi.org/10.2174/1574888X14666190429143734 (2019).
Siebel, C. & Lendahl, U. Notch signaling in development, tissue homeostasis, and disease. Physiol. Rev. 97, 1235–1294 (2017).
Haltiwanger, R. S. et al. In Essentials of Glycobiology (eds A. Varki et al.) 155–164 (2022).
Tsukamoto, Y. et al. Glycoproteomics of NOTCH1 EGF repeat fragments overexpressed with different glycosyltransferases in HEK293T cells reveals insights into O-GlcNAcylation of NOTCH1. Glycobiology 32, 616–628. https://doi.org/10.1093/glycob/cwac015 (2022).
Varshney, S. & Stanley, P. Multiple roles for O-glycans in Notch signalling. FEBS Lett. 592, 3819–3834. https://doi.org/10.1002/1873-3468.13251 (2018).
Pandey, A., Niknejad, N. & Jafar-Nejad, H. Multifaceted regulation of Notch signaling by glycosylation. Glycobiology 31, 8–28. https://doi.org/10.1093/glycob/cwaa049 (2021).
Matsumoto, K., Luther, K. B. & Haltiwanger, R. S. Diseases related to Notch glycosylation. Mol. Aspects Med. 20, 100938. https://doi.org/10.1016/j.mam.2020.100938 (2020).
Rampal, R., Luther, K. B. & Haltiwanger, R. S. Notch signaling in normal and disease States: Possible therapies related to glycosylation. Curr. Mol. Med. 7, 427–445. https://doi.org/10.2174/156652407780831593 (2007).
Shi, S. et al. The threonine that carries fucose, but not fucose, is required for Cripto to facilitate Nodal signaling. J. Biol. Chem. 282, 20133–20141. https://doi.org/10.1074/jbc.M702593200 (2007).
Luca, V. C. et al. Structural biology. Structural basis for Notch1 engagement of Delta-like 4. Science 347, 847–853. https://doi.org/10.1126/science.1261093 (2015).
Luca, V. C. et al. Notch-Jagged complex structure implicates a catch bond in tuning ligand sensitivity. Science 355, 1320–1324. https://doi.org/10.1126/science.aaf9739 (2017).
Varshney, S. et al. A modifier in the 129S2/SvPasCrl genome is responsible for the viability of Notch1[12f/12f] mice. Bmc Dev. Biol. 19, 1–11. https://doi.org/10.1186/s12861-019-0199-3 (2019).
Guilmeau, S. et al. Intestinal deletion of Pofut1 in the mouse inactivates notch signaling and causes enterocolitis. Gastroenterology 135(849–860), e841-846. https://doi.org/10.1053/j.gastro.2008.05.050 (2008).
Murthy, P. K. L. et al. Radical and lunatic fringes modulate notch ligands to support mammalian intestinal homeostasis. Elife 7, e35710. https://doi.org/10.7554/eLife.35710 (2018).
Pellegrinet, L. et al. Dll1-and Dll4-mediated notch signaling are required for homeostasis of intestinal stem cells. Gastroenterology 140, 1230–1240 (2011).
Riccio, O. et al. Loss of intestinal crypt progenitor cells owing to inactivation of both Notch1 and Notch2 is accompanied by derepression of CDK inhibitors p27Kip1 and p57Kip2. EMBO Rep. 9, 377–383 (2008).
van Es, J. H., de Geest, N., van de Born, M., Clevers, H. & Hassan, B. A. Intestinal stem cells lacking the Math1 tumour suppressor are refractory to Notch inhibitors. Nat. Commun. 1, 18. https://doi.org/10.1038/ncomms1017 (2010).
Sakaidani, Y. et al. O-linked-N-acetylglucosamine on extracellular protein domains mediates epithelial cell-matrix interactions. Nat. Commun. 2, 583. https://doi.org/10.1038/ncomms1591 (2011).
Sawaguchi, S. et al. O-GlcNAc on NOTCH1 EGF repeats regulates ligand-induced Notch signaling and vascular development in mammals. Elife 6, e24419. https://doi.org/10.7554/eLife.24419 (2017).
Harvey, B. M. & Haltiwanger, R. S. Regulation of notch function by O-glycosylation. Adv. Exp. Med. Biol. 1066, 59–78. https://doi.org/10.1007/978-3-319-89512-3_4 (2018).
Ogawa, M., Senoo, Y., Ikeda, K., Takeuchi, H. & Okajima, T. Structural divergence in O-GlcNAc glycans displayed on epidermal growth factor-like repeats of mammalian notch1. Molecules 23, 1745. https://doi.org/10.3390/molecules23071745 (2018).
Saiki, W., Ma, C., Okajima, T. & Takeuchi, H. Current views on the roles of O-glycosylation in controlling notch-ligand interactions. Biomolecules 11, 309. https://doi.org/10.3390/biom11020309 (2021).
Chen, W. & Stanley, P. Five Lec1 CHO cell mutants have distinct Mgat1 gene mutations that encode truncated N-acetylglucosaminyltransferase I. Glycobiology 13, 43–50. https://doi.org/10.1093/glycob/cwg003 (2003).
Tashima, Y. & Stanley, P. Antibodies that detect O-linked beta-D-N-acetylglucosamine on the extracellular domain of cell surface glycoproteins. J. Biol. Chem. 289, 11132–11142. https://doi.org/10.1074/jbc.M113.492512 (2014).
Tian, H. et al. Opposing activities of Notch and Wnt signaling regulate intestinal stem cells and gut homeostasis. Cell Rep. 11, 33–42 (2015).
Carulli, A. J. et al. Notch receptor regulation of intestinal stem cell homeostasis and crypt regeneration. Dev. Biol. 402, 98–108. https://doi.org/10.1016/j.ydbio.2015.03.012 (2015).
Fernandez-Valdivia, R. et al. Regulation of mammalian Notch signaling and embryonic development by the protein O-glucosyltransferase Rumi. Development 138, 1925–1934 (2011).
Takeuchi, H. et al. O-Glycosylation modulates the stability of epidermal growth factor-like repeats and thereby regulates Notch trafficking. J. Biol. Chem. 292, 15964–15973. https://doi.org/10.1074/jbc.M117.800102 (2017).
Shaheen, R. et al. Mutations in EOGT confirm the genetic heterogeneity of autosomal-recessive Adams–Oliver syndrome. Am. J. Hum. Genet. 92, 598–604. https://doi.org/10.1016/j.ajhg.2013.02.012 (2013).
Schroder, K. C. et al. Adams–Oliver syndrome caused by mutations of the EOGT gene. Am. J. Med. Genet. A 179, 2246–2251. https://doi.org/10.1002/ajmg.a.61313 (2019).
Gao, J. et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 6, l1. https://doi.org/10.1126/scisignal.2004088 (2013).
Matsumoto, K. et al. Fringe GlcNAc-transferases differentially extend O-fucose on endogenous NOTCH1 in mouse activated T cells. J. Biol. Chem. 298, 102064. https://doi.org/10.1016/j.jbc.2022.102064 (2022).
Choi, J. et al. Intestinal stem cell aging at single-cell resolution: Functional perturbations alter cell developmental trajectory reversed by gerotherapeutics. BioRxiv https://doi.org/10.1101/2022.08.31.505869 (2022).
Wang, Y. et al. Uncontrolled angiogenic precursor expansion causes coronary artery anomalies in mice lacking Pofut1. Nat. Commun. 8, 1–15 (2017).
Stahl, M. et al. Roles of Pofut1 and O-fucose in mammalian Notch signaling. J. Biol. Chem. 283, 13638–13651. https://doi.org/10.1074/jbc.M802027200 (2008).
Shi, S. & Stanley, P. Protein O-fucosyltransferase 1 is an essential component of Notch signaling pathways. Proc. Natl. Acad. Sci. USA 100, 5234–5239. https://doi.org/10.1073/pnas.0831126100 (2003).
Acknowledgements
We thank the Histology and Comparative Pathology Core, Albert Einstein College of Medicine, for sectioning and staining fixed intestine samples. We thank Andrea Briceno and Hillary Guzik, Analytical Imaging Facility, Albert Einstein College of Medicine, for scanning histology slides using a 3D Histech P250 High-Capacity Slide Scanner purchased with NIH SIG #1S10OD019961-01. We are grateful to Jinghang Zhang and Aodengtuya of the Flow Cytometry Core, Albert Einstein College, for support in designing and executing flow cytometry experiments. We also thank Subha Sundaram for overall technical assistance.
Funding
This work was funded by National Institutes of Health Grants R01 GM106417 (PS), R01 CA229216 (LHA), and partially supported by the Albert Einstein Cancer center Grant PO1 CA13330.
Author information
Authors and Affiliations
Contributions
Conceptualization, P.S.; methodology, N.M., S.V., P.S., J.C., L.H.A.; investigation, N.M., S.V.; analysis, N.M., S.V., J.C., L.H.A., P.S.; resources, P.S., L.H.A.; writing first draft, N.M., P.S.; review and editing, P.S., N.M., L.H.A., J.C.; visualization, N.M., J.C.; funding acquisition, P.S., L.H.A.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Nauman, M., Varshney, S., Choi, J. et al. EOGT enables residual Notch signaling in mouse intestinal cells lacking POFUT1. Sci Rep 13, 17473 (2023). https://doi.org/10.1038/s41598-023-44509-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-44509-5
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
